{"title":"Compound heterozygous mutations in three Chinese patients of Segawa syndrome and their treatment outcomes","authors":"Jie Zhang, Yaxin Huang, Yulei Hu, Bing Bai","doi":"10.1002/jdn.10328","DOIUrl":null,"url":null,"abstract":"<p>Segawa syndrome is a rare autosomal recessive form of dopa-responsive dystonia resulting from <i>TH</i> gene dysfunction. Patients typically exhibit symptoms such as generalized dystonia, rigidity, tremors, infantile Parkinsonism, and pseudo-spastic paraplegia. Levodopa is often an effective treatment. Due to its rarity, high heterogeneity, and poorly understood pathological mutation and phenotype spectrums, as well as genotype–phenotype and genotype-treatment outcome correlations, Segawa syndrome poses diagnostic and therapeutic challenges. In our study, through clinical and molecular analyses of three Chinese Segawa patients, we re-evaluated the pathogenicity of a <i>TH</i> mutation (c.880G>C;p.G294R) previously categorized as “Conflicting classifications of pathogenicity” in ClinVar. Also, we summarized the clinical phenotypes of all reported Segawa syndrome cases until 2023 and compared them with our patients. We identified a novel phenotype, “cafe-au-lait macules,” not previously observed in Segawa patients. Additionally, we discussed the correlation between specific genotypes and phenotypes, as well as genotypes and treatment outcomes of our three cases. Our findings aim to enhance the understanding of Segawa syndrome, contributing to improved diagnosis and treatment approaches in the future.</p>","PeriodicalId":13914,"journal":{"name":"International Journal of Developmental Neuroscience","volume":"84 4","pages":"305-313"},"PeriodicalIF":1.7000,"publicationDate":"2024-04-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Developmental Neuroscience","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jdn.10328","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"DEVELOPMENTAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
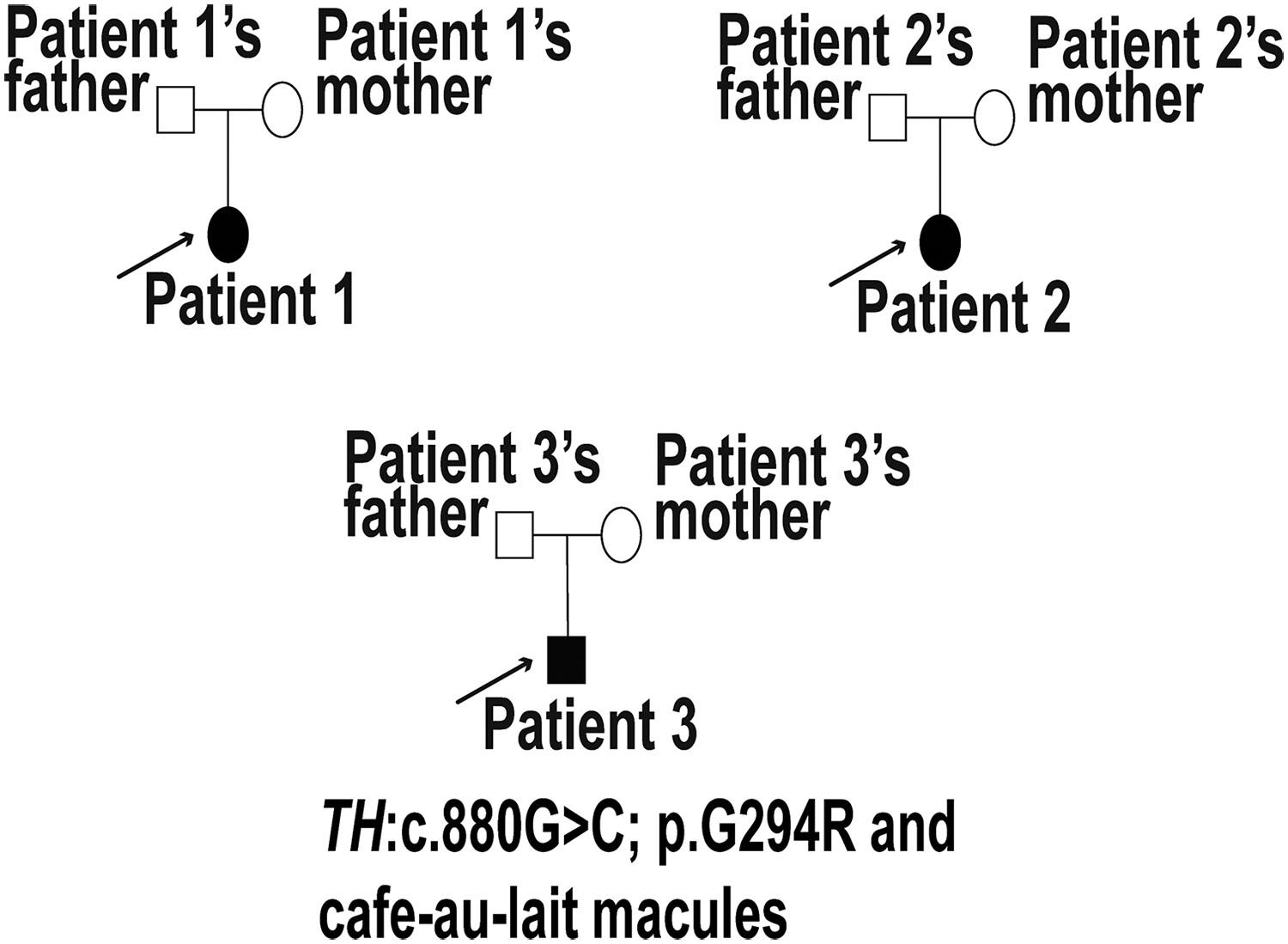
Segawa syndrome is a rare autosomal recessive form of dopa-responsive dystonia resulting from TH gene dysfunction. Patients typically exhibit symptoms such as generalized dystonia, rigidity, tremors, infantile Parkinsonism, and pseudo-spastic paraplegia. Levodopa is often an effective treatment. Due to its rarity, high heterogeneity, and poorly understood pathological mutation and phenotype spectrums, as well as genotype–phenotype and genotype-treatment outcome correlations, Segawa syndrome poses diagnostic and therapeutic challenges. In our study, through clinical and molecular analyses of three Chinese Segawa patients, we re-evaluated the pathogenicity of a TH mutation (c.880G>C;p.G294R) previously categorized as “Conflicting classifications of pathogenicity” in ClinVar. Also, we summarized the clinical phenotypes of all reported Segawa syndrome cases until 2023 and compared them with our patients. We identified a novel phenotype, “cafe-au-lait macules,” not previously observed in Segawa patients. Additionally, we discussed the correlation between specific genotypes and phenotypes, as well as genotypes and treatment outcomes of our three cases. Our findings aim to enhance the understanding of Segawa syndrome, contributing to improved diagnosis and treatment approaches in the future.
期刊介绍:
International Journal of Developmental Neuroscience publishes original research articles and critical review papers on all fundamental and clinical aspects of nervous system development, renewal and regeneration, as well as on the effects of genetic and environmental perturbations of brain development and homeostasis leading to neurodevelopmental disorders and neurological conditions. Studies describing the involvement of stem cells in nervous system maintenance and disease (including brain tumours), stem cell-based approaches for the investigation of neurodegenerative diseases, roles of neuroinflammation in development and disease, and neuroevolution are also encouraged. Investigations using molecular, cellular, physiological, genetic and epigenetic approaches in model systems ranging from simple invertebrates to human iPSC-based 2D and 3D models are encouraged, as are studies using experimental models that provide behavioural or evolutionary insights. The journal also publishes Special Issues dealing with topics at the cutting edge of research edited by Guest Editors appointed by the Editor in Chief. A major aim of the journal is to facilitate the transfer of fundamental studies of nervous system development, maintenance, and disease to clinical applications. The journal thus intends to disseminate valuable information for both biologists and physicians. International Journal of Developmental Neuroscience is owned and supported by The International Society for Developmental Neuroscience (ISDN), an organization of scientists interested in advancing developmental neuroscience research in the broadest sense.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


