Rare genetic variation in fibronectin 1 (FN1) protects against APOEε4 in Alzheimer’s disease
Abstract
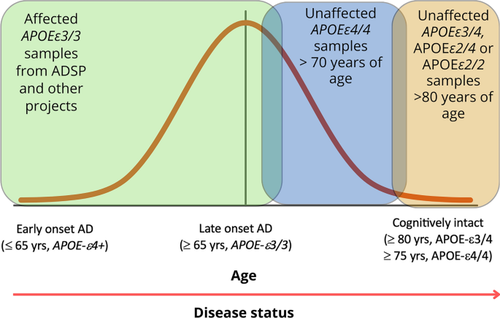
The risk of developing Alzheimer’s disease (AD) significantly increases in individuals carrying the APOEε4 allele. Elderly cognitively healthy individuals with APOEε4 also exist, suggesting the presence of cellular mechanisms that counteract the pathological effects of APOEε4; however, these mechanisms are unknown. We hypothesized that APOEε4 carriers without dementia might carry genetic variations that could protect them from developing APOEε4-mediated AD pathology. To test this, we leveraged whole-genome sequencing (WGS) data in the National Institute on Aging Alzheimer's Disease Family Based Study (NIA-AD FBS), Washington Heights/Inwood Columbia Aging Project (WHICAP), and Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA) cohorts and identified potentially protective variants segregating exclusively among unaffected APOEε4 carriers. In homozygous unaffected carriers above 70 years old, we identified 510 rare coding variants. Pathway analysis of the genes harboring these variants showed significant enrichment in extracellular matrix (ECM)-related processes, suggesting protective effects of functional modifications in ECM proteins. We prioritized two genes that were highly represented in the ECM-related gene ontology terms, (FN1) and collagen type VI alpha 2 chain (COL6A2) and are known to be expressed at the blood–brain barrier (BBB), for postmortem validation and in vivo functional studies. An independent analysis in a large cohort of 7185 APOEε4 homozygous carriers found that rs140926439 variant in FN1 was protective of AD (OR = 0.29; 95% CI [0.11, 0.78], P = 0.014) and delayed age at onset of disease by 3.37 years (95% CI [0.42, 6.32], P = 0.025). The FN1 and COL6A2 protein levels were increased at the BBB in APOEε4 carriers with AD. Brain expression of cognitively unaffected homozygous APOEε4 carriers had significantly lower FN1 deposition and less reactive gliosis compared to homozygous APOEε4 carriers with AD, suggesting that FN1 might be a downstream driver of APOEε4-mediated AD-related pathology and cognitive decline. To validate our findings, we used zebrafish models with loss-of-function (LOF) mutations in fn1b—the ortholog for human FN1. We found that fibronectin LOF reduced gliosis, enhanced gliovascular remodeling, and potentiated the microglial response, suggesting that pathological accumulation of FN1 could impair toxic protein clearance, which is ameliorated with FN1 LOF. Our study suggests that vascular deposition of FN1 is related to the pathogenicity of APOEε4, and LOF variants in FN1 may reduce APOEε4-related AD risk, providing novel clues to potential therapeutic interventions targeting the ECM to mitigate AD risk.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


