Inge Kinnart, Liselot Manders, Thibaut Heyninck, Dorien Imberechts, Roman Praschberger, Nils Schoovaerts, Catherine Verfaillie, Patrik Verstreken, Wim Vandenberghe
{"title":"Elevated α-synuclein levels inhibit mitophagic flux","authors":"Inge Kinnart, Liselot Manders, Thibaut Heyninck, Dorien Imberechts, Roman Praschberger, Nils Schoovaerts, Catherine Verfaillie, Patrik Verstreken, Wim Vandenberghe","doi":"10.1038/s41531-024-00696-0","DOIUrl":null,"url":null,"abstract":"<p>The pathogenic effect of <i>SNCA</i> gene multiplications indicates that elevation of wild-type α-synuclein levels is sufficient to cause Parkinson’s disease (PD). Mitochondria have been proposed to be a major target of α-synuclein-induced damage. PINK1/parkin/DJ-1-mediated mitophagy is a defense strategy that allows cells to selectively eliminate severely damaged mitochondria. Here, we quantified mitophagic flux and non-mitochondrial autophagic flux in three models of increased α-synuclein expression: 1/<i>Drosophila melanogaster</i> that transgenically express human wild-type and mutant α-synuclein in flight muscle; 2/human skin fibroblasts transfected with α-synuclein or β-synuclein; and 3/human induced pluripotent stem cell (iPSC)-derived neurons carrying an extra copy of wild-type <i>SNCA</i> under control of a doxycycline-inducible promoter, allowing titratable α-synuclein upregulation. In each model, elevated α-synuclein levels potently suppressed mitophagic flux, while non-mitochondrial autophagy was preserved. In human neurons, a twofold increase in wild-type α-synuclein was already sufficient to induce this effect. PINK1 and parkin activation and mitochondrial translocation of DJ-1 after mitochondrial depolarization were not affected by α-synuclein upregulation. Overexpression of the actin-severing protein cofilin or treatment with CK666, an inhibitor of the actin-related protein 2/3 (Arp2/3) complex, rescued mitophagy in neurons with increased α-synuclein, suggesting that excessive actin network stabilization mediated the mitophagy defect. In conclusion, elevated α-synuclein levels inhibit mitophagic flux. Disruption of actin dynamics may play a key role in this effect.</p>","PeriodicalId":19706,"journal":{"name":"NPJ Parkinson's Disease","volume":null,"pages":null},"PeriodicalIF":6.7000,"publicationDate":"2024-04-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Parkinson's Disease","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41531-024-00696-0","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
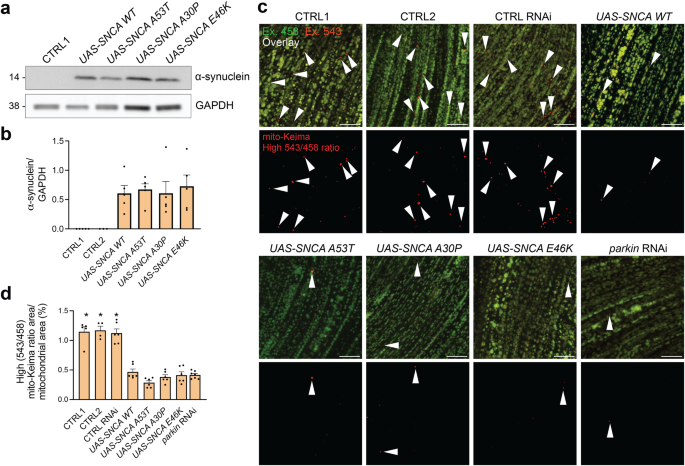
The pathogenic effect of SNCA gene multiplications indicates that elevation of wild-type α-synuclein levels is sufficient to cause Parkinson’s disease (PD). Mitochondria have been proposed to be a major target of α-synuclein-induced damage. PINK1/parkin/DJ-1-mediated mitophagy is a defense strategy that allows cells to selectively eliminate severely damaged mitochondria. Here, we quantified mitophagic flux and non-mitochondrial autophagic flux in three models of increased α-synuclein expression: 1/Drosophila melanogaster that transgenically express human wild-type and mutant α-synuclein in flight muscle; 2/human skin fibroblasts transfected with α-synuclein or β-synuclein; and 3/human induced pluripotent stem cell (iPSC)-derived neurons carrying an extra copy of wild-type SNCA under control of a doxycycline-inducible promoter, allowing titratable α-synuclein upregulation. In each model, elevated α-synuclein levels potently suppressed mitophagic flux, while non-mitochondrial autophagy was preserved. In human neurons, a twofold increase in wild-type α-synuclein was already sufficient to induce this effect. PINK1 and parkin activation and mitochondrial translocation of DJ-1 after mitochondrial depolarization were not affected by α-synuclein upregulation. Overexpression of the actin-severing protein cofilin or treatment with CK666, an inhibitor of the actin-related protein 2/3 (Arp2/3) complex, rescued mitophagy in neurons with increased α-synuclein, suggesting that excessive actin network stabilization mediated the mitophagy defect. In conclusion, elevated α-synuclein levels inhibit mitophagic flux. Disruption of actin dynamics may play a key role in this effect.
期刊介绍:
npj Parkinson's Disease is a comprehensive open access journal that covers a wide range of research areas related to Parkinson's disease. It publishes original studies in basic science, translational research, and clinical investigations. The journal is dedicated to advancing our understanding of Parkinson's disease by exploring various aspects such as anatomy, etiology, genetics, cellular and molecular physiology, neurophysiology, epidemiology, and therapeutic development. By providing free and immediate access to the scientific and Parkinson's disease community, npj Parkinson's Disease promotes collaboration and knowledge sharing among researchers and healthcare professionals.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


