Greta Grassmann, Mattia Miotto, Fausta Desantis, Lorenzo Di Rienzo, Gian Gaetano Tartaglia, Annalisa Pastore, Giancarlo Ruocco, Michele Monti and Edoardo Milanetti*,
{"title":"Computational Approaches to Predict Protein–Protein Interactions in Crowded Cellular Environments","authors":"Greta Grassmann, Mattia Miotto, Fausta Desantis, Lorenzo Di Rienzo, Gian Gaetano Tartaglia, Annalisa Pastore, Giancarlo Ruocco, Michele Monti and Edoardo Milanetti*, ","doi":"10.1021/acs.chemrev.3c00550","DOIUrl":null,"url":null,"abstract":"<p >Investigating protein–protein interactions is crucial for understanding cellular biological processes because proteins often function within molecular complexes rather than in isolation. While experimental and computational methods have provided valuable insights into these interactions, they often overlook a critical factor: the crowded cellular environment. This environment significantly impacts protein behavior, including structural stability, diffusion, and ultimately the nature of binding. In this review, we discuss theoretical and computational approaches that allow the modeling of biological systems to guide and complement experiments and can thus significantly advance the investigation, and possibly the predictions, of protein–protein interactions in the crowded environment of cell cytoplasm. We explore topics such as statistical mechanics for lattice simulations, hydrodynamic interactions, diffusion processes in high-viscosity environments, and several methods based on molecular dynamics simulations. By synergistically leveraging methods from biophysics and computational biology, we review the state of the art of computational methods to study the impact of molecular crowding on protein–protein interactions and discuss its potential revolutionizing effects on the characterization of the human interactome.</p>","PeriodicalId":32,"journal":{"name":"Chemical Reviews","volume":"124 7","pages":"3932–3977"},"PeriodicalIF":55.8000,"publicationDate":"2024-03-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.chemrev.3c00550","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Reviews","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.chemrev.3c00550","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
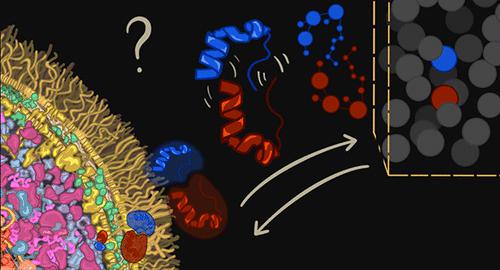
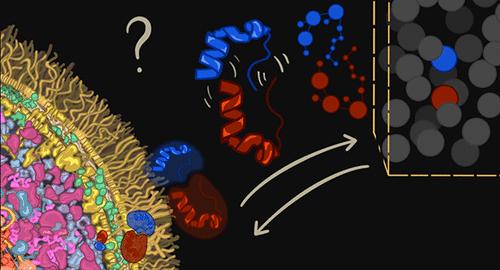
Investigating protein–protein interactions is crucial for understanding cellular biological processes because proteins often function within molecular complexes rather than in isolation. While experimental and computational methods have provided valuable insights into these interactions, they often overlook a critical factor: the crowded cellular environment. This environment significantly impacts protein behavior, including structural stability, diffusion, and ultimately the nature of binding. In this review, we discuss theoretical and computational approaches that allow the modeling of biological systems to guide and complement experiments and can thus significantly advance the investigation, and possibly the predictions, of protein–protein interactions in the crowded environment of cell cytoplasm. We explore topics such as statistical mechanics for lattice simulations, hydrodynamic interactions, diffusion processes in high-viscosity environments, and several methods based on molecular dynamics simulations. By synergistically leveraging methods from biophysics and computational biology, we review the state of the art of computational methods to study the impact of molecular crowding on protein–protein interactions and discuss its potential revolutionizing effects on the characterization of the human interactome.
期刊介绍:
Chemical Reviews is a highly regarded and highest-ranked journal covering the general topic of chemistry. Its mission is to provide comprehensive, authoritative, critical, and readable reviews of important recent research in organic, inorganic, physical, analytical, theoretical, and biological chemistry.
Since 1985, Chemical Reviews has also published periodic thematic issues that focus on a single theme or direction of emerging research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


