A mediation analysis framework based on variance component to remove genetic confounding effect
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
Abstract
Identification of pleiotropy at the single nucleotide polymorphism (SNP) level provides valuable insights into shared genetic signals among phenotypes. One approach to study these signals is through mediation analysis, which dissects the total effect of a SNP on the outcome into a direct effect and an indirect effect through a mediator. However, estimated effects from mediation analysis can be confounded by the genetic correlation between phenotypes, leading to inaccurate results. To address this confounding effect in the context of genetic mediation analysis, we propose a restricted-maximum-likelihood (REML)-based mediation analysis framework called REML-mediation, which can be applied to either individual-level or summary statistics data. Simulations demonstrated that REML-mediation provides unbiased estimates of the true cross-trait causal effect, assuming certain assumptions, albeit with a slightly inflated standard error compared to traditional linear regression. To validate the effectiveness of REML-mediation, we applied it to UK Biobank data and analyzed several mediator-outcome trait pairs along with their corresponding sets of pleiotropic SNPs. REML-mediation successfully identified and corrected for genetic confounding effects in these trait pairs, with correction magnitudes ranging from 7% to 39%. These findings highlight the presence of genetic confounding effects in cross-trait epidemiological studies and underscore the importance of accounting for them in data analysis.
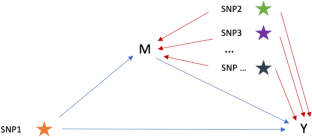
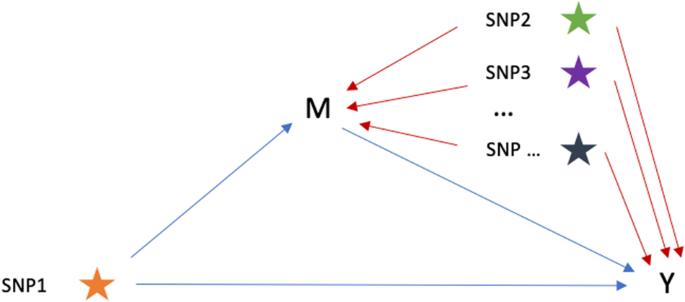
基于变异成分的中介分析框架可消除遗传混杂效应。
在单核苷酸多态性(SNP)水平上识别多效性,为了解表型之间共享的遗传信号提供了宝贵的见解。研究这些信号的一种方法是进行中介分析,将 SNP 对结果的总效应分解为直接效应和通过中介产生的间接效应。然而,中介分析的估计效应可能会受到表型之间遗传相关性的干扰,导致结果不准确。为了解决遗传中介分析中的这种混杂效应,我们提出了一种基于受限最大似然法(REML)的中介分析框架,称为 REML-中介,它既可应用于个体水平数据,也可应用于汇总统计数据。模拟结果表明,与传统的线性回归相比,REML-中介分析尽管标准误差略有增大,但能在一定假设条件下提供真实的跨性状因果效应的无偏估计值。为了验证 REML 中介法的有效性,我们将其应用于英国生物库数据,分析了几对中介因子-结果性状及其相应的多效 SNP。REML-mediation 成功识别并校正了这些性状对中的遗传混杂效应,校正幅度从 7% 到 39% 不等。这些发现凸显了跨性状流行病学研究中遗传混杂效应的存在,并强调了在数据分析中考虑这些效应的重要性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


