Haikuo Li, Dian Li, Nicolas Ledru, Qiao Xuanyuan, Haojia Wu, Amish Asthana, Lori N Byers, Stefan G Tullius, Giuseppe Orlando, Sushrut S Waikar, Benjamin D Humphreys
{"title":"Transcriptomic, epigenomic, and spatial metabolomic cell profiling redefines regional human kidney anatomy.","authors":"Haikuo Li, Dian Li, Nicolas Ledru, Qiao Xuanyuan, Haojia Wu, Amish Asthana, Lori N Byers, Stefan G Tullius, Giuseppe Orlando, Sushrut S Waikar, Benjamin D Humphreys","doi":"10.1016/j.cmet.2024.02.015","DOIUrl":null,"url":null,"abstract":"<p><p>A large-scale multimodal atlas that includes major kidney regions is lacking. Here, we employed simultaneous high-throughput single-cell ATAC/RNA sequencing (SHARE-seq) and spatially resolved metabolomics to profile 54 human samples from distinct kidney anatomical regions. We generated transcriptomes of 446,267 cells and chromatin accessibility profiles of 401,875 cells and developed a package to analyze 408,218 spatially resolved metabolomes. We find that the same cell type, including thin limb, thick ascending limb loop of Henle and principal cells, display distinct transcriptomic, chromatin accessibility, and metabolomic signatures, depending on anatomic location. Surveying metabolism-associated gene profiles revealed non-overlapping metabolic signatures between nephron segments and dysregulated lipid metabolism in diseased proximal tubule (PT) cells. Integrating multimodal omics with clinical data identified PLEKHA1 as a disease marker, and its in vitro knockdown increased gene expression in PT differentiation, suggesting possible pathogenic roles. This study highlights previously underrepresented cellular heterogeneity underlying the human kidney anatomy.</p>","PeriodicalId":93927,"journal":{"name":"Cell metabolism","volume":" ","pages":"1105-1125.e10"},"PeriodicalIF":30.9000,"publicationDate":"2024-05-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11081846/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell metabolism","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.cmet.2024.02.015","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/3/20 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
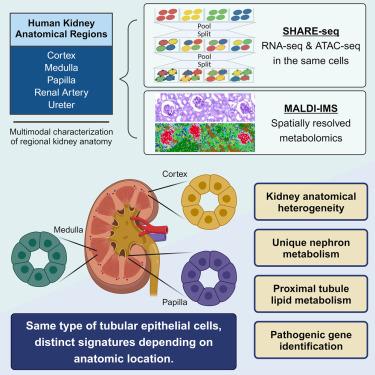
A large-scale multimodal atlas that includes major kidney regions is lacking. Here, we employed simultaneous high-throughput single-cell ATAC/RNA sequencing (SHARE-seq) and spatially resolved metabolomics to profile 54 human samples from distinct kidney anatomical regions. We generated transcriptomes of 446,267 cells and chromatin accessibility profiles of 401,875 cells and developed a package to analyze 408,218 spatially resolved metabolomes. We find that the same cell type, including thin limb, thick ascending limb loop of Henle and principal cells, display distinct transcriptomic, chromatin accessibility, and metabolomic signatures, depending on anatomic location. Surveying metabolism-associated gene profiles revealed non-overlapping metabolic signatures between nephron segments and dysregulated lipid metabolism in diseased proximal tubule (PT) cells. Integrating multimodal omics with clinical data identified PLEKHA1 as a disease marker, and its in vitro knockdown increased gene expression in PT differentiation, suggesting possible pathogenic roles. This study highlights previously underrepresented cellular heterogeneity underlying the human kidney anatomy.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


