Huiwen Deng, Cui He, Alexandra Z. Worden, Jun Gong
{"title":"Employing a triple metabarcoding approach to differentiate active, dormant and dead microeukaryotes in sediments","authors":"Huiwen Deng, Cui He, Alexandra Z. Worden, Jun Gong","doi":"10.1111/1462-2920.16615","DOIUrl":null,"url":null,"abstract":"<p>Microbial communities are commonly characterised through the metabarcoding of environmental DNA. This DNA originates from both viable (including dormant and active) and dead organisms, leading to recent efforts to distinguish between these states. In this study, we further these approaches by distinguishing not only between viable and dead cells but also between dormant and actively growing cells. This is achieved by sequencing both rRNA and rDNA, in conjunction with propidium monoazide cross-linked rDNA, to partition the active, dormant and relic fractions in environmental samples. We apply this method to characterise the diversity and assemblage structure of these fractions of microeukaryotes in intertidal sediments during a wet-dry-rewet incubation cycle. Our findings indicate that a significant proportion of microeukaryotic phylotypes detected in the total rDNA pools originate from dormant and relic microeukaryotes in the sediments, both in terms of richness (dormant, 13 ± 2%; relic, 47 ± 5%) and read abundance (dormant, 20 ± 7%; relic, 14 ± 5%). The richness and sequence proportion of dormant microeukaryotes notably increase during the transition from wet to dry conditions. Statistical analyses suggest that the dynamics of diversity and assemblage structure across different activity fractions are influenced by various environmental drivers. Our strategy offers a versatile approach that can be adapted to characterise other microbes in a wide range of environments.</p>","PeriodicalId":11898,"journal":{"name":"Environmental microbiology","volume":"26 3","pages":""},"PeriodicalIF":4.3000,"publicationDate":"2024-03-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Environmental microbiology","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/1462-2920.16615","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
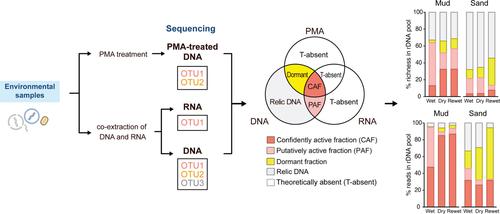
Microbial communities are commonly characterised through the metabarcoding of environmental DNA. This DNA originates from both viable (including dormant and active) and dead organisms, leading to recent efforts to distinguish between these states. In this study, we further these approaches by distinguishing not only between viable and dead cells but also between dormant and actively growing cells. This is achieved by sequencing both rRNA and rDNA, in conjunction with propidium monoazide cross-linked rDNA, to partition the active, dormant and relic fractions in environmental samples. We apply this method to characterise the diversity and assemblage structure of these fractions of microeukaryotes in intertidal sediments during a wet-dry-rewet incubation cycle. Our findings indicate that a significant proportion of microeukaryotic phylotypes detected in the total rDNA pools originate from dormant and relic microeukaryotes in the sediments, both in terms of richness (dormant, 13 ± 2%; relic, 47 ± 5%) and read abundance (dormant, 20 ± 7%; relic, 14 ± 5%). The richness and sequence proportion of dormant microeukaryotes notably increase during the transition from wet to dry conditions. Statistical analyses suggest that the dynamics of diversity and assemblage structure across different activity fractions are influenced by various environmental drivers. Our strategy offers a versatile approach that can be adapted to characterise other microbes in a wide range of environments.
期刊介绍:
Environmental Microbiology provides a high profile vehicle for publication of the most innovative, original and rigorous research in the field. The scope of the Journal encompasses the diversity of current research on microbial processes in the environment, microbial communities, interactions and evolution and includes, but is not limited to, the following:
the structure, activities and communal behaviour of microbial communities
microbial community genetics and evolutionary processes
microbial symbioses, microbial interactions and interactions with plants, animals and abiotic factors
microbes in the tree of life, microbial diversification and evolution
population biology and clonal structure
microbial metabolic and structural diversity
microbial physiology, growth and survival
microbes and surfaces, adhesion and biofouling
responses to environmental signals and stress factors
modelling and theory development
pollution microbiology
extremophiles and life in extreme and unusual little-explored habitats
element cycles and biogeochemical processes, primary and secondary production
microbes in a changing world, microbially-influenced global changes
evolution and diversity of archaeal and bacterial viruses
new technological developments in microbial ecology and evolution, in particular for the study of activities of microbial communities, non-culturable microorganisms and emerging pathogens

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


