Nikhil Gadewal , Abhiram Natu , Siddhartha Sen , Sukanya Rauniyar , Virupaksha Bastikar , Sanjay Gupta
{"title":"Integrative epigenome-transcriptome analysis unravels cancer-specific over-expressed genes potentially regulating immune microenvironment in clear cell renal cell carcinoma","authors":"Nikhil Gadewal , Abhiram Natu , Siddhartha Sen , Sukanya Rauniyar , Virupaksha Bastikar , Sanjay Gupta","doi":"10.1016/j.bbagen.2024.130596","DOIUrl":null,"url":null,"abstract":"<div><h3>Background</h3><p>Clear cell Renal Cell Carcinoma (ccRCC) is the frequently diagnosed histological life-threatening tumor subtype in the urinary system. Integrating multi-omics data is emerging as a tool to provide a comprehensive view of biology and disease for better therapeutic interventions.</p></div><div><h3>Method</h3><p>We have integrated freely available ccRCC data sets of genome-wide DNA methylome, transcriptome, and active histone modification marks, H3K27ac, H3K4me1, and H3K4me3 specific ChIP-seq data to screen genes with higher expression. Further, these genes were filtered based on their effect on survival upon alteration in expression.</p></div><div><h3>Results</h3><p>The six multi-omics-based identified genes, RUNX1, MSC, ADA, TREML1, TGFA, and VWF, showed higher expression with enrichment of active histone marks and hypomethylated CpG in ccRCC. In continuation, the identified genes were validated by an independent dataset and showed a correlation with nodal and metastatic status. Furthermore, gene ontology and pathway analysis revealed that immune-related pathways are activated in ccRCC patients.</p></div><div><h3>Conclusions</h3><p>The network analysis of six overexpressed genes suggests their potential role in an immunosuppressive environment, leading to tumor progression and poor prognosis. Our study shows that the multi-omics approach helps unravel complex biology for patient subtyping and proposes combination strategies with epi-drugs for more precise immunotherapy in ccRCC.</p></div>","PeriodicalId":8800,"journal":{"name":"Biochimica et biophysica acta. General subjects","volume":null,"pages":null},"PeriodicalIF":2.8000,"publicationDate":"2024-03-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biochimica et biophysica acta. General subjects","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0304416524000394","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background
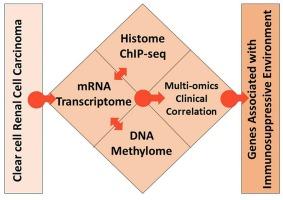
Clear cell Renal Cell Carcinoma (ccRCC) is the frequently diagnosed histological life-threatening tumor subtype in the urinary system. Integrating multi-omics data is emerging as a tool to provide a comprehensive view of biology and disease for better therapeutic interventions.
Method
We have integrated freely available ccRCC data sets of genome-wide DNA methylome, transcriptome, and active histone modification marks, H3K27ac, H3K4me1, and H3K4me3 specific ChIP-seq data to screen genes with higher expression. Further, these genes were filtered based on their effect on survival upon alteration in expression.
Results
The six multi-omics-based identified genes, RUNX1, MSC, ADA, TREML1, TGFA, and VWF, showed higher expression with enrichment of active histone marks and hypomethylated CpG in ccRCC. In continuation, the identified genes were validated by an independent dataset and showed a correlation with nodal and metastatic status. Furthermore, gene ontology and pathway analysis revealed that immune-related pathways are activated in ccRCC patients.
Conclusions
The network analysis of six overexpressed genes suggests their potential role in an immunosuppressive environment, leading to tumor progression and poor prognosis. Our study shows that the multi-omics approach helps unravel complex biology for patient subtyping and proposes combination strategies with epi-drugs for more precise immunotherapy in ccRCC.
期刊介绍:
BBA General Subjects accepts for submission either original, hypothesis-driven studies or reviews covering subjects in biochemistry and biophysics that are considered to have general interest for a wide audience. Manuscripts with interdisciplinary approaches are especially encouraged.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


