Leena Putzeys, Laura Wicke, Maarten Boon, Vera van Noort, Jörg Vogel, Rob Lavigne
{"title":"Refining the transcriptional landscapes for distinct clades of virulent phages infecting <i>Pseudomonas aeruginosa</i>.","authors":"Leena Putzeys, Laura Wicke, Maarten Boon, Vera van Noort, Jörg Vogel, Rob Lavigne","doi":"10.1093/femsml/uqae002","DOIUrl":null,"url":null,"abstract":"<p><p>The introduction of high-throughput sequencing has resulted in a surge of available bacteriophage genomes, unveiling their tremendous genomic diversity. However, our current understanding of the complex transcriptional mechanisms that dictate their gene expression during infection is limited to a handful of model phages. Here, we applied ONT-cappable-seq to reveal the transcriptional architecture of six different clades of virulent phages infecting <i>Pseudomonas aeruginosa</i>. This long-read microbial transcriptomics approach is tailored to globally map transcription start and termination sites, transcription units, and putative RNA-based regulators on dense phage genomes. Specifically, the full-length transcriptomes of LUZ19, LUZ24, 14-1, YuA, PAK_P3, and giant phage phiKZ during early, middle, and late infection were collectively charted. Beyond pinpointing traditional promoter and terminator elements and transcription units, these transcriptional profiles provide insights in transcriptional attenuation and splicing events and allow straightforward validation of Group I intron activity. In addition, ONT-cappable-seq data can guide genome-wide discovery of novel regulatory element candidates, including noncoding RNAs and riboswitches. This work substantially expands the number of annotated phage-encoded transcriptional elements identified to date, shedding light on the intricate and diverse gene expression regulation mechanisms in <i>Pseudomonas</i> phages, which can ultimately be sourced as tools for biotechnological applications in phage and bacterial engineering.</p>","PeriodicalId":74189,"journal":{"name":"microLife","volume":"5 ","pages":"uqae002"},"PeriodicalIF":0.0000,"publicationDate":"2024-02-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10914365/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"microLife","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/femsml/uqae002","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
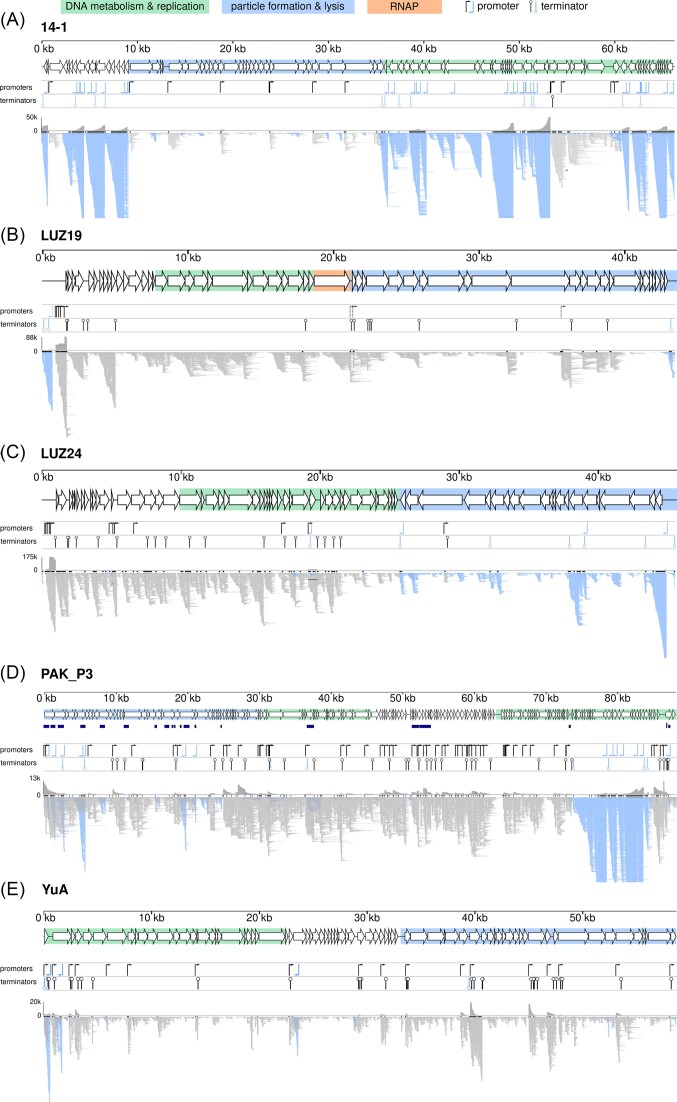
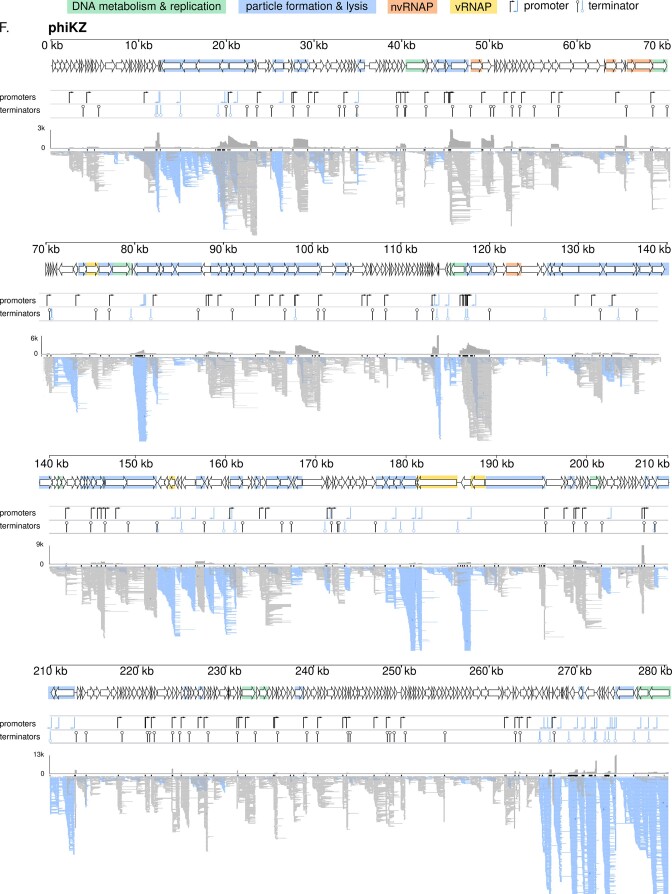
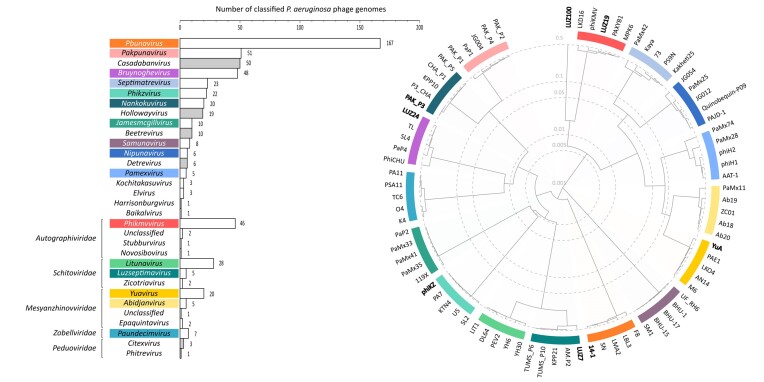
The introduction of high-throughput sequencing has resulted in a surge of available bacteriophage genomes, unveiling their tremendous genomic diversity. However, our current understanding of the complex transcriptional mechanisms that dictate their gene expression during infection is limited to a handful of model phages. Here, we applied ONT-cappable-seq to reveal the transcriptional architecture of six different clades of virulent phages infecting Pseudomonas aeruginosa. This long-read microbial transcriptomics approach is tailored to globally map transcription start and termination sites, transcription units, and putative RNA-based regulators on dense phage genomes. Specifically, the full-length transcriptomes of LUZ19, LUZ24, 14-1, YuA, PAK_P3, and giant phage phiKZ during early, middle, and late infection were collectively charted. Beyond pinpointing traditional promoter and terminator elements and transcription units, these transcriptional profiles provide insights in transcriptional attenuation and splicing events and allow straightforward validation of Group I intron activity. In addition, ONT-cappable-seq data can guide genome-wide discovery of novel regulatory element candidates, including noncoding RNAs and riboswitches. This work substantially expands the number of annotated phage-encoded transcriptional elements identified to date, shedding light on the intricate and diverse gene expression regulation mechanisms in Pseudomonas phages, which can ultimately be sourced as tools for biotechnological applications in phage and bacterial engineering.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


