Shidong Xi, Tania Nguyen, Struan Murray, Phil Lorenz, Jane Mellor
{"title":"Size fractionated NET-Seq reveals a conserved architecture of transcription units around yeast genes.","authors":"Shidong Xi, Tania Nguyen, Struan Murray, Phil Lorenz, Jane Mellor","doi":"10.1002/yea.3931","DOIUrl":null,"url":null,"abstract":"<p><p>Genomes from yeast to humans are subject to pervasive transcription. A single round of pervasive transcription is sufficient to alter local chromatin conformation, nucleosome dynamics and gene expression, but is hard to distinguish from background signals. Size fractionated native elongating transcript sequencing (sfNET-Seq) was developed to precisely map nascent transcripts independent of expression levels. RNAPII-associated nascent transcripts are fractionation into different size ranges before library construction. When anchored to the transcription start sites (TSS) of annotated genes, the combined pattern of the output metagenes gives the expected reference pattern. Bioinformatic pattern matching to the reference pattern identified 9542 transcription units in Saccharomyces cerevisiae, of which 47% are coding and 53% are noncoding. In total, 3113 (33%) are unannotated noncoding transcription units. Anchoring all transcription units to the TSS or polyadenylation site (PAS) of annotated genes reveals distinctive architectures of linked pairs of divergent transcripts approximately 200nt apart. The Reb1 transcription factor is enriched 30nt downstream of the PAS only when an upstream (TSS -60nt with respect to PAS) noncoding transcription unit co-occurs with a downstream (TSS +150nt) coding transcription unit and acts to limit levels of upstream antisense transcripts. The potential for extensive transcriptional interference is evident from low abundance unannotated transcription units with variable TSS (median -240nt) initiating within a 500nt window upstream of, and transcribing over, the promoters of protein-coding genes. This study confirms a highly interleaved yeast genome with different types of transcription units altering the chromatin landscape in distinctive ways, with the potential to exert extensive regulatory control.</p>","PeriodicalId":23870,"journal":{"name":"Yeast","volume":" ","pages":"222-241"},"PeriodicalIF":2.6000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Yeast","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1002/yea.3931","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/3/3 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
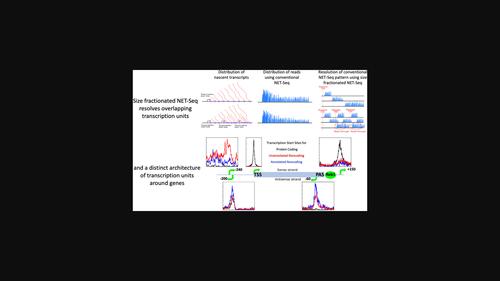
Genomes from yeast to humans are subject to pervasive transcription. A single round of pervasive transcription is sufficient to alter local chromatin conformation, nucleosome dynamics and gene expression, but is hard to distinguish from background signals. Size fractionated native elongating transcript sequencing (sfNET-Seq) was developed to precisely map nascent transcripts independent of expression levels. RNAPII-associated nascent transcripts are fractionation into different size ranges before library construction. When anchored to the transcription start sites (TSS) of annotated genes, the combined pattern of the output metagenes gives the expected reference pattern. Bioinformatic pattern matching to the reference pattern identified 9542 transcription units in Saccharomyces cerevisiae, of which 47% are coding and 53% are noncoding. In total, 3113 (33%) are unannotated noncoding transcription units. Anchoring all transcription units to the TSS or polyadenylation site (PAS) of annotated genes reveals distinctive architectures of linked pairs of divergent transcripts approximately 200nt apart. The Reb1 transcription factor is enriched 30nt downstream of the PAS only when an upstream (TSS -60nt with respect to PAS) noncoding transcription unit co-occurs with a downstream (TSS +150nt) coding transcription unit and acts to limit levels of upstream antisense transcripts. The potential for extensive transcriptional interference is evident from low abundance unannotated transcription units with variable TSS (median -240nt) initiating within a 500nt window upstream of, and transcribing over, the promoters of protein-coding genes. This study confirms a highly interleaved yeast genome with different types of transcription units altering the chromatin landscape in distinctive ways, with the potential to exert extensive regulatory control.
期刊介绍:
Yeast publishes original articles and reviews on the most significant developments of research with unicellular fungi, including innovative methods of broad applicability. It is essential reading for those wishing to keep up to date with this rapidly moving field of yeast biology.
Topics covered include: biochemistry and molecular biology; biodiversity and taxonomy; biotechnology; cell and developmental biology; ecology and evolution; genetics and genomics; metabolism and physiology; pathobiology; synthetic and systems biology; tools and resources

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


