Christoph Hofmann , Marjan Aghajani , Cecily D. Alcock , Erik A. Blackwood , Clara Sandmann , Nicole Herzog , Julia Groß , Lars Plate , R. Luke Wiseman , Randal J. Kaufman , Hugo A. Katus , Tobias Jakobi , Mirko Völkers , Christopher C. Glembotski , Shirin Doroudgar
{"title":"ATF6 protects against protein misfolding during cardiac hypertrophy","authors":"Christoph Hofmann , Marjan Aghajani , Cecily D. Alcock , Erik A. Blackwood , Clara Sandmann , Nicole Herzog , Julia Groß , Lars Plate , R. Luke Wiseman , Randal J. Kaufman , Hugo A. Katus , Tobias Jakobi , Mirko Völkers , Christopher C. Glembotski , Shirin Doroudgar","doi":"10.1016/j.yjmcc.2024.02.001","DOIUrl":null,"url":null,"abstract":"<div><p>Cardiomyocytes activate the unfolded protein response (UPR) transcription factor ATF6 during pressure overload-induced hypertrophic growth. The UPR is thought to increase ER protein folding capacity and maintain proteostasis. ATF6 deficiency during pressure overload leads to heart failure, suggesting that ATF6 protects against myocardial dysfunction by preventing protein misfolding. However, conclusive evidence that ATF6 prevents toxic protein misfolding during cardiac hypertrophy is still pending. Here, we found that activation of the UPR, including ATF6, is a common response to pathological cardiac hypertrophy in mice. ATF6 KO mice failed to induce sufficient levels of UPR target genes in response to chronic isoproterenol infusion or transverse aortic constriction (TAC), resulting in impaired cardiac growth. To investigate the effects of ATF6 on protein folding, the accumulation of poly-ubiquitinated proteins as well as soluble amyloid oligomers were directly quantified in hypertrophied hearts of WT and ATF6 KO mice. Whereas only low levels of protein misfolding was observed in WT hearts after TAC, ATF6 KO mice accumulated increased quantities of misfolded protein, which was associated with impaired myocardial function. Collectively, the data suggest that ATF6 plays a critical adaptive role during cardiac hypertrophy by protecting against protein misfolding.</p></div>","PeriodicalId":16402,"journal":{"name":"Journal of molecular and cellular cardiology","volume":"189 ","pages":"Pages 12-24"},"PeriodicalIF":4.9000,"publicationDate":"2024-02-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular and cellular cardiology","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S002228282400018X","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CARDIAC & CARDIOVASCULAR SYSTEMS","Score":null,"Total":0}
引用次数: 0
Abstract
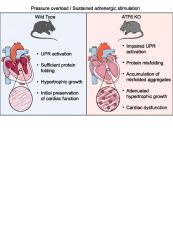
Cardiomyocytes activate the unfolded protein response (UPR) transcription factor ATF6 during pressure overload-induced hypertrophic growth. The UPR is thought to increase ER protein folding capacity and maintain proteostasis. ATF6 deficiency during pressure overload leads to heart failure, suggesting that ATF6 protects against myocardial dysfunction by preventing protein misfolding. However, conclusive evidence that ATF6 prevents toxic protein misfolding during cardiac hypertrophy is still pending. Here, we found that activation of the UPR, including ATF6, is a common response to pathological cardiac hypertrophy in mice. ATF6 KO mice failed to induce sufficient levels of UPR target genes in response to chronic isoproterenol infusion or transverse aortic constriction (TAC), resulting in impaired cardiac growth. To investigate the effects of ATF6 on protein folding, the accumulation of poly-ubiquitinated proteins as well as soluble amyloid oligomers were directly quantified in hypertrophied hearts of WT and ATF6 KO mice. Whereas only low levels of protein misfolding was observed in WT hearts after TAC, ATF6 KO mice accumulated increased quantities of misfolded protein, which was associated with impaired myocardial function. Collectively, the data suggest that ATF6 plays a critical adaptive role during cardiac hypertrophy by protecting against protein misfolding.
期刊介绍:
The Journal of Molecular and Cellular Cardiology publishes work advancing knowledge of the mechanisms responsible for both normal and diseased cardiovascular function. To this end papers are published in all relevant areas. These include (but are not limited to): structural biology; genetics; proteomics; morphology; stem cells; molecular biology; metabolism; biophysics; bioengineering; computational modeling and systems analysis; electrophysiology; pharmacology and physiology. Papers are encouraged with both basic and translational approaches. The journal is directed not only to basic scientists but also to clinical cardiologists who wish to follow the rapidly advancing frontiers of basic knowledge of the heart and circulation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


