{"title":"Bioequivalence of a New Pediatric Paracetamol Oral Suspension Compared With a Marketed Formulation in Healthy Adults: A Randomized, Open-Label Study","authors":"Jiri Grim MD, PhD , Marianna Armogida MD , Preeti Kachroo BAMS, MD(AM) , Kamran Siddiqui MBBS, MBA , Mauro Cavinato PhD , Mako Araga MS","doi":"10.1016/j.curtheres.2024.100734","DOIUrl":null,"url":null,"abstract":"<div><h3>Background</h3><p>A new oral paracetamol formulation with the same paracetamol quantity (24 mg/mL) as a marketed formulation but with finer active ingredient particle size and lower amounts of maltitol (5.85 g/dose in the test formulation vs 7.25 g/dose in the reference formulation) and sorbitol (2.4 g/dose vs 2.83 g/dose) was developed.</p></div><div><h3>Objective</h3><p>Establish the bioequivalence of the new pediatric formulation (test treatment) compared with the marketed formulation (reference treatment).</p></div><div><h3>Methods</h3><p>This Phase I, open-label trial assigned healthy adult volunteers to a single 42-mL (1 g para-cetamol) dose of test or reference treatment. Participants received both treatments in a randomized order separated by a 72-hour washout period. The primary endpoints were AUC<sub>0–tlast</sub> (AUC vs time curve from time 0 to last measurable sampling timepoint), C<sub>max</sub>, and t<sub>max</sub>. Safety assessments included adverse event, clinical laboratory, and physical examination data.</p></div><div><h3>Results</h3><p>Thirty-five participants were randomized and treated. The study population was 42.9% women (57.1% men) with a median age of 30 years; most participants were non-Hispanic White. Mean C<sub>max</sub> values were comparable between test and reference products, with a median t<sub>max</sub> of 1.00 hour for both. The test/reference ratios (%) (90% CI) for AUC<sub>0–tlast</sub> and C<sub>max</sub> were 98.69% (96.46, 100.97) and 100.73% (95.63, 106.10), respectively. There were no adverse events or deaths.</p></div><div><h3>Conclusions</h3><p>The new paracetamol formulation is bioequivalent to the marketed formulation.</p></div>","PeriodicalId":10920,"journal":{"name":"Current Therapeutic Research-clinical and Experimental","volume":"100 ","pages":"Article 100734"},"PeriodicalIF":1.5000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0011393X24000043/pdfft?md5=47ca70cb77c917b984528fd3d13b1234&pid=1-s2.0-S0011393X24000043-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Therapeutic Research-clinical and Experimental","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0011393X24000043","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
Background
A new oral paracetamol formulation with the same paracetamol quantity (24 mg/mL) as a marketed formulation but with finer active ingredient particle size and lower amounts of maltitol (5.85 g/dose in the test formulation vs 7.25 g/dose in the reference formulation) and sorbitol (2.4 g/dose vs 2.83 g/dose) was developed.
Objective
Establish the bioequivalence of the new pediatric formulation (test treatment) compared with the marketed formulation (reference treatment).
Methods
This Phase I, open-label trial assigned healthy adult volunteers to a single 42-mL (1 g para-cetamol) dose of test or reference treatment. Participants received both treatments in a randomized order separated by a 72-hour washout period. The primary endpoints were AUC0–tlast (AUC vs time curve from time 0 to last measurable sampling timepoint), Cmax, and tmax. Safety assessments included adverse event, clinical laboratory, and physical examination data.
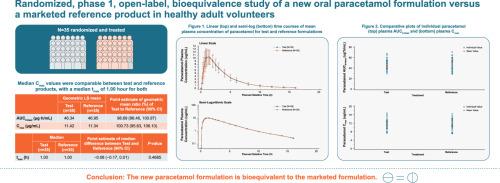
Results
Thirty-five participants were randomized and treated. The study population was 42.9% women (57.1% men) with a median age of 30 years; most participants were non-Hispanic White. Mean Cmax values were comparable between test and reference products, with a median tmax of 1.00 hour for both. The test/reference ratios (%) (90% CI) for AUC0–tlast and Cmax were 98.69% (96.46, 100.97) and 100.73% (95.63, 106.10), respectively. There were no adverse events or deaths.
Conclusions
The new paracetamol formulation is bioequivalent to the marketed formulation.
期刊介绍:
We also encourage the submission of manuscripts presenting preclinical and very preliminary research that may stimulate further investigation of potentially relevant findings, as well as in-depth review articles on specific therapies or disease states, and applied health delivery or pharmacoeconomics.
CTR encourages and supports the submission of manuscripts describing:
• Interventions designed to understand or improve human health, disease treatment or disease prevention;
• Studies that focus on problems that are uncommon in resource-rich countries;
• Research that is "under-published" because of limited access to monetary resources such as English language support and Open Access fees (CTR offers deeply discounted English language editing);
• Republication of articles previously published in non-English journals (eg, evidence-based guidelines) which could be useful if translated into English;
• Preclinical and clinical product development studies that are not pursued for further investigation based upon early phase results.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


