{"title":"A theoretical study for the linear free energy relationship of CH bond activation and the role of the axial ligand in cytochrome P450 model complexes","authors":"Soobin Kwon, Yun-Cheol Choi, Yongho Kim","doi":"10.1002/bkcs.12819","DOIUrl":null,"url":null,"abstract":"<p>Hydrogen abstraction is essential for C<span></span>H bond activation by Compound I in cytochrome P450 and is influenced by various factors, including spin states, bond dissociation energies of the C<span></span>H and FeO<span></span>H bonds, axial ligands, and quantum mechanical tunneling. The role of axial ligands has been extensively studied, but it is still not fully understood. To explore their role, we used density functional theory calculations to determine whether a linear free energy relationship is established for the hydrogen transfer reaction, according to changes in axial ligands. The B3LYP* functional exhibits a strong linear correlation, but the slopes are inconsistent with the characteristics of the transition state. Natural bond orbital analysis reveals no direct orbital interaction between axial ligands and the reaction center of hydrogen transfer. The electron-donating orbitals of the axial ligands weaken the Fe<span></span>O bond, lowering the energy barrier, but they do not directly participate in the intrinsic hydrogen transfer. During the reaction, the Fe<span></span>O bond length increases significantly before the hydrogen transfer itself, generating an asynchronous shift in the bond orders, and most of the activation energy is used for the increase in the Fe<span></span>O bond rather than the hydrogen transfer itself. This study may explain why there is no apparent correlation between the rate constants and the Fe<span></span>O bond strength.</p>","PeriodicalId":54252,"journal":{"name":"Bulletin of the Korean Chemical Society","volume":"45 3","pages":"284-292"},"PeriodicalIF":1.7000,"publicationDate":"2024-02-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bulletin of the Korean Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/bkcs.12819","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
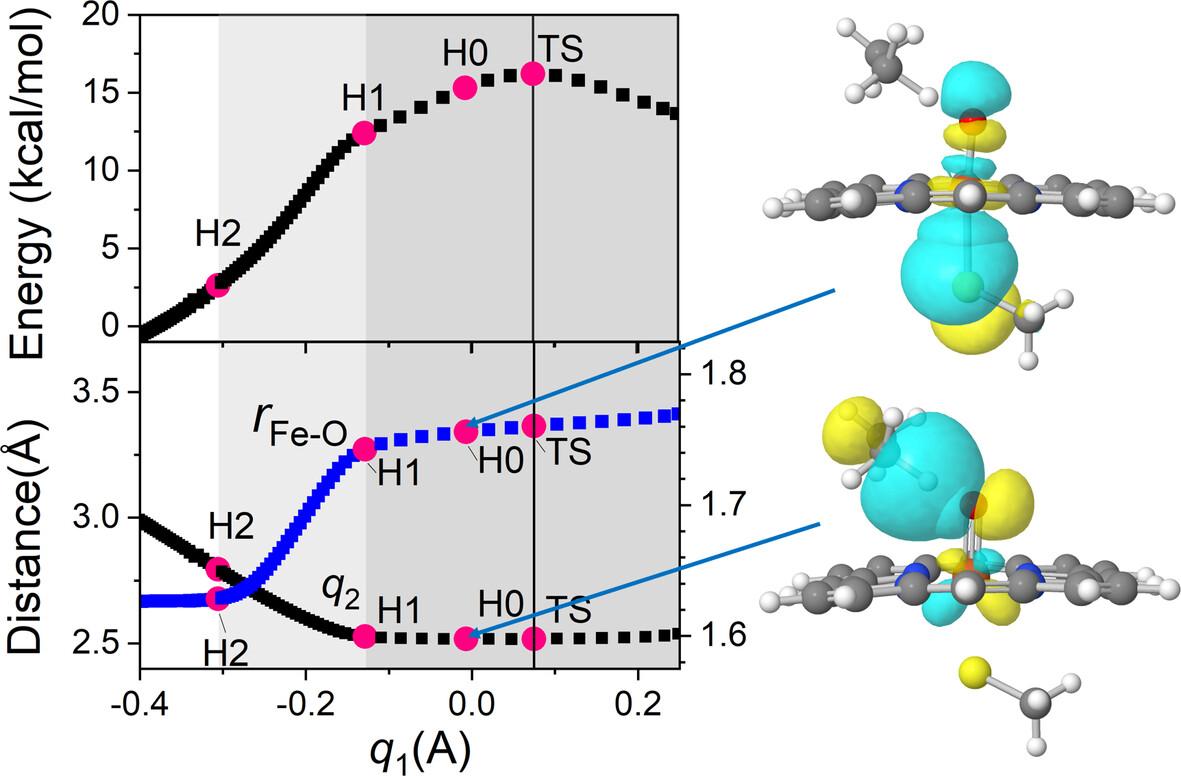
Hydrogen abstraction is essential for CH bond activation by Compound I in cytochrome P450 and is influenced by various factors, including spin states, bond dissociation energies of the CH and FeOH bonds, axial ligands, and quantum mechanical tunneling. The role of axial ligands has been extensively studied, but it is still not fully understood. To explore their role, we used density functional theory calculations to determine whether a linear free energy relationship is established for the hydrogen transfer reaction, according to changes in axial ligands. The B3LYP* functional exhibits a strong linear correlation, but the slopes are inconsistent with the characteristics of the transition state. Natural bond orbital analysis reveals no direct orbital interaction between axial ligands and the reaction center of hydrogen transfer. The electron-donating orbitals of the axial ligands weaken the FeO bond, lowering the energy barrier, but they do not directly participate in the intrinsic hydrogen transfer. During the reaction, the FeO bond length increases significantly before the hydrogen transfer itself, generating an asynchronous shift in the bond orders, and most of the activation energy is used for the increase in the FeO bond rather than the hydrogen transfer itself. This study may explain why there is no apparent correlation between the rate constants and the FeO bond strength.
期刊介绍:
The Bulletin of the Korean Chemical Society is an official research journal of the Korean Chemical Society. It was founded in 1980 and reaches out to the chemical community worldwide. It is strictly peer-reviewed and welcomes Accounts, Communications, Articles, and Notes written in English. The scope of the journal covers all major areas of chemistry: analytical chemistry, electrochemistry, industrial chemistry, inorganic chemistry, life-science chemistry, macromolecular chemistry, organic synthesis, non-synthetic organic chemistry, physical chemistry, and materials chemistry.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


