Sequencing and characterizing short tandem repeats in the human genome
IF 39.1
1区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
Abstract
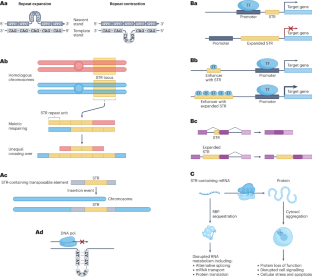
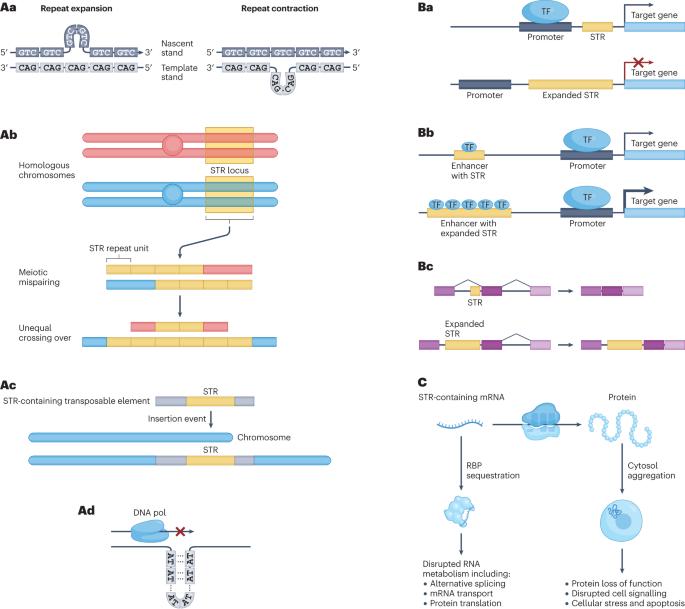
Short tandem repeats (STRs) are highly polymorphic sequences throughout the human genome that are composed of repeated copies of a 1–6-bp motif. Over 1 million variable STR loci are known, some of which regulate gene expression and influence complex traits, such as height. Moreover, variants in at least 60 STR loci cause genetic disorders, including Huntington disease and fragile X syndrome. Accurately identifying and genotyping STR variants is challenging, in particular mapping short reads to repetitive regions and inferring expanded repeat lengths. Recent advances in sequencing technology and computational tools for STR genotyping from sequencing data promise to help overcome this challenge and solve genetically unresolved cases and the ‘missing heritability’ of polygenic traits. Here, we compare STR genotyping methods, analytical tools and their applications to understand the effect of STR variation on health and disease. We identify emergent opportunities to refine genotyping and quality-control approaches as well as to integrate STRs into variant-calling workflows and large cohort analyses. This Review describes tools and approaches for characterizing short tandem repeats in the human genome from whole-genome sequencing data. Furthermore, the authors discuss how these recent developments have helped to better understand the effect of short tandem repeats on human health and disease.
人类基因组中短串联重复序列的测序和特征描述。
短串联重复序列(STR)是遍布人类基因组的高度多态性序列,由 1-6-bp 主题的重复拷贝组成。目前已知的可变 STR 位点超过 100 万个,其中一些位点调节基因表达并影响复杂的性状,如身高。此外,至少有 60 个 STR 位点的变异会导致遗传疾病,包括亨廷顿病和脆性 X 综合征。准确鉴定和基因分型 STR 变异是一项挑战,尤其是将短读数映射到重复区域和推断扩大的重复长度。测序技术和计算工具在从测序数据中进行 STR 基因分型方面的最新进展有望帮助克服这一挑战,解决遗传学上的未决病例和多基因性状的 "缺失遗传性 "问题。在这里,我们比较了 STR 基因分型方法、分析工具及其应用,以了解 STR 变异对健康和疾病的影响。我们发现了改进基因分型和质量控制方法以及将 STR 纳入变异调用工作流程和大型队列分析的新机遇。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊
Nature Reviews Genetics
生物-遗传学
CiteScore
57.40
自引率
0.50%
发文量
113
审稿时长
6-12 weeks
期刊介绍:
At Nature Reviews Genetics, our goal is to be the leading source of reviews and commentaries for the scientific communities we serve. We are dedicated to publishing authoritative articles that are easily accessible to our readers. We believe in enhancing our articles with clear and understandable figures, tables, and other display items. Our aim is to provide an unparalleled service to authors, referees, and readers, and we are committed to maximizing the usefulness and impact of each article we publish.
Within our journal, we publish a range of content including Research Highlights, Comments, Reviews, and Perspectives that are relevant to geneticists and genomicists. With our broad scope, we ensure that the articles we publish reach the widest possible audience.
As part of the Nature Reviews portfolio of journals, we strive to uphold the high standards and reputation associated with this esteemed collection of publications.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


