Genetic etiology of truncus arteriosus excluding 22q11.2 deletion syndrome and identification of c.1617del, a prevalent variant in TMEM260, in the Japanese population
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
Abstract
Truncus Arteriosus (TA) is a congenital heart disease characterized by a single common blood vessel emerging from the right and left ventricles instead of the main pulmonary artery and aorta. TA accounts for 4% of all critical congenital heart diseases. The most common cause of TA is 22q11.2 deletion syndrome, accounting for 12–35% of all TA cases. However, no major causes of TA other than 22q11.2 deletion have been reported. We performed whole-genome sequencing of 11 Japanese patients having TA without 22q11.2 deletion. Among five patients, we identified pathogenic variants in TMEM260; the biallelic loss-of-function variants of which have recently been associated with structural heart defects and renal anomalies syndrome (SHDRA). In one patient, we identified a de novo pathogenic variant in GATA6, and in another patient, we identified a de novo probably pathogenic variant in NOTCH1. Notably, we identified a prevalent variant in TMEM260 (ENST00000261556.6), c.1617del (p.Trp539Cysfs*9), in 8/22 alleles among the 11 patients. The c.1617del variant was estimated to occur approximately 23 kiloyears ago. Based on the allele frequency of the c.1617del variant in the Japanese population (0.36%), approximately 26% of Japanese patients afflicted with TA could harbor homozygous c.1617del variants. This study highlights TMEM260, especially c.1617del, as a major genetic cause of TA in the Japanese population.
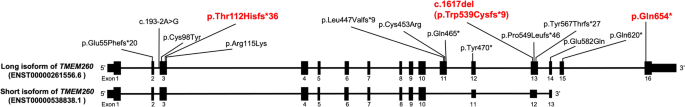
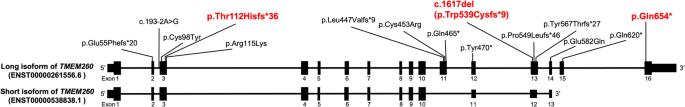
日本人群中排除 22q11.2 缺失综合征的动脉导管未闭的遗传学病因,以及 TMEM260 的流行变异 c.1617del 的鉴定。
动脉导管未闭(TA)是一种先天性心脏病,其特点是左右心室有一根共同的血管,而不是主肺动脉和主动脉。在所有危重先天性心脏病中,TA 占 4%。TA最常见的病因是22q11.2缺失综合征,占所有TA病例的12-35%。然而,除22q11.2缺失外,尚无其他导致TA的主要病因的报道。我们对 11 名没有 22q11.2 缺失的日本 TA 患者进行了全基因组测序。在五名患者中,我们发现了 TMEM260 的致病变体;其双偶功能缺失变体最近与结构性心脏缺陷和肾脏异常综合征(SHDRA)有关。在一名患者身上,我们发现了 GATA6 的从头致病变异;在另一名患者身上,我们发现了 NOTCH1 的从头可能致病变异。值得注意的是,我们在11名患者中的8/22个等位基因中发现了TMEM260(ENST00000261556.6)的一个流行变异c.1617del(p.Trp539Cysfs*9)。据估计,c.1617del 变异发生在大约 23 千年前。根据c.1617del变异在日本人群中的等位基因频率(0.36%),大约26%的日本TA患者可能携带同源c.1617del变异。这项研究表明,TMEM260,尤其是c.1617del,是日本人群中TA的主要遗传病因。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


