Taeeun Kim, Ahwon Lee, Stephan Ahn, Jae Sung Park, Sin Soo Jeun, Youn Soo Lee
{"title":"Comprehensive Molecular Genetic Analysis in Glioma Patients by Next Generation Sequencing.","authors":"Taeeun Kim, Ahwon Lee, Stephan Ahn, Jae Sung Park, Sin Soo Jeun, Youn Soo Lee","doi":"10.14791/btrt.2023.0036","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Glioma is caused by multiple genomic alterations. The evolving classification of gliomas emphasizes the significance of molecular testing. Next generation sequencing (NGS) offers the assessment of parallel combinations of multiple genetic alterations and identifying actionable mutations that guide treatment. This study comprehensively analyzed glioma patients using multi-gene NGS panels, providing powerful insights to inform diagnostic classification and targeted therapies.</p><p><strong>Methods: </strong>We conducted a targeted panel-based NGS analysis on formalin-fixed and paraffin-embedded nucleic acids extracted from a total of 147 glioma patients. These samples underwent amplicon capture-based library preparation and sequenced using the Oncomine Comprehensive Assay platform. The resulting sequencing data were then analyzed using the bioinformatics tools.</p><p><strong>Results: </strong>A total of 301 mutations, were found in 132 out of 147 tumors (89.8%). These mutations were in 68 different genes. In 62 tumor samples (42.2%), copy number variations (CNVs) with gene amplifications occurred in 25 genes. Moreover, 25 tumor samples (17.0%) showed gene fusions in 6 genes and intragenic deletion in a gene. Our analysis identified actionable targets in several genes, including 11 with mutations, 8 with CNVs, and 3 with gene fusions and intragenic deletion. These findings could impact FDA-approved therapies, NCCN guideline-based treatments, and clinical trials.</p><p><strong>Conclusion: </strong>We analyzed precisely diagnosing the classification of gliomas, detailing the frequency and co-occurrence of genetic alterations and identifying genetic alterations with potential therapeutic targets by NGS-based molecular analysis. The high-throughput NGS analysis is an efficient and powerful tool to comprehensively support molecular testing in neurooncology.</p>","PeriodicalId":72453,"journal":{"name":"Brain tumor research and treatment","volume":"12 1","pages":"23-39"},"PeriodicalIF":0.0000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10864139/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain tumor research and treatment","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.14791/btrt.2023.0036","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Glioma is caused by multiple genomic alterations. The evolving classification of gliomas emphasizes the significance of molecular testing. Next generation sequencing (NGS) offers the assessment of parallel combinations of multiple genetic alterations and identifying actionable mutations that guide treatment. This study comprehensively analyzed glioma patients using multi-gene NGS panels, providing powerful insights to inform diagnostic classification and targeted therapies.
Methods: We conducted a targeted panel-based NGS analysis on formalin-fixed and paraffin-embedded nucleic acids extracted from a total of 147 glioma patients. These samples underwent amplicon capture-based library preparation and sequenced using the Oncomine Comprehensive Assay platform. The resulting sequencing data were then analyzed using the bioinformatics tools.
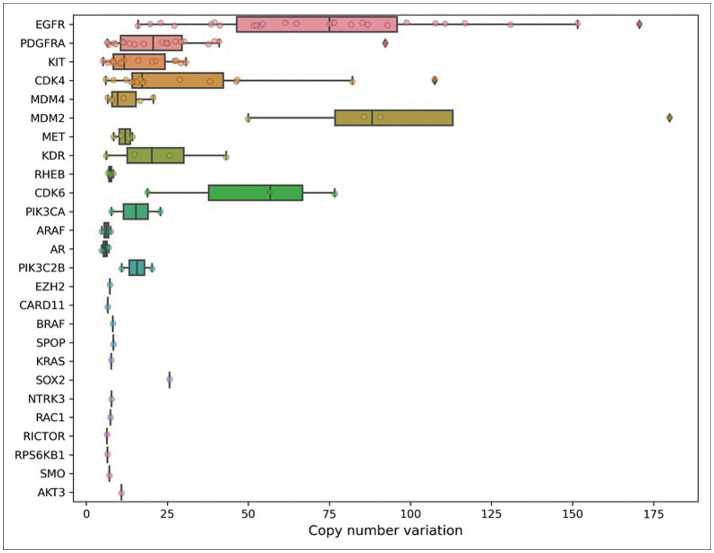
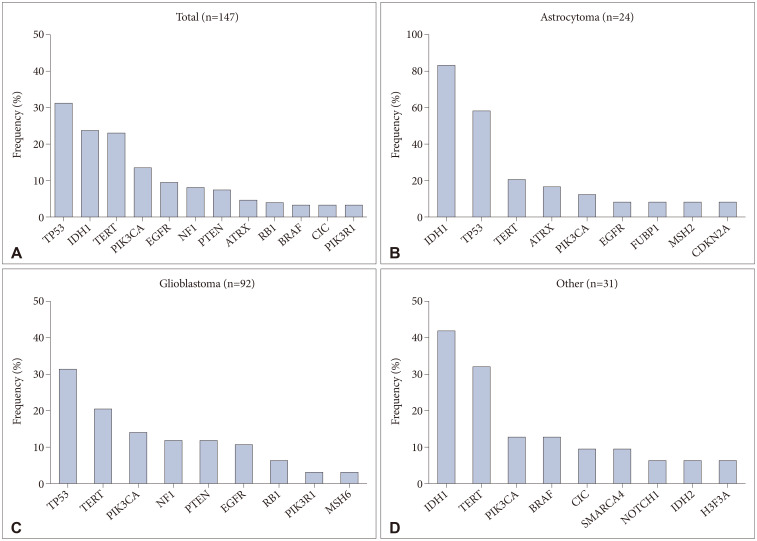
Results: A total of 301 mutations, were found in 132 out of 147 tumors (89.8%). These mutations were in 68 different genes. In 62 tumor samples (42.2%), copy number variations (CNVs) with gene amplifications occurred in 25 genes. Moreover, 25 tumor samples (17.0%) showed gene fusions in 6 genes and intragenic deletion in a gene. Our analysis identified actionable targets in several genes, including 11 with mutations, 8 with CNVs, and 3 with gene fusions and intragenic deletion. These findings could impact FDA-approved therapies, NCCN guideline-based treatments, and clinical trials.
Conclusion: We analyzed precisely diagnosing the classification of gliomas, detailing the frequency and co-occurrence of genetic alterations and identifying genetic alterations with potential therapeutic targets by NGS-based molecular analysis. The high-throughput NGS analysis is an efficient and powerful tool to comprehensively support molecular testing in neurooncology.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


