{"title":"The SMA Modifier Plastin 3 Targets Cell Membrane-Associated Proteins in Motoneurons.","authors":"Sibylle Jablonka, Natascha Schäfer","doi":"10.1177/26331055241226623","DOIUrl":null,"url":null,"abstract":"<p><p>Loss of the <i>Survival Motor Neuron (SMN)</i> gene inevitably leads to spinal muscular atrophy (SMA), one of the most common fatal neuromuscular diseases in children with FDA and EMA approved therapies. However, the cellular mechanisms leading to neuromuscular junction (NMJ) dysfunction due to impaired Ca<sup>2+</sup> homeostasis in the presynaptic compartment remain largely unexplained. In the last decade, the so-called SMA modifiers have gained attention. The F-actin bundler Plastin 3 (PLS3) is one of them and counteracts neurotransmission defects, including altered vesicle endocytosis, in Smn-deficient NMJs. Properly bundled F-actin is the basis for the translocation and arrangement of transmembrane proteins at the cell surface. Our recently published data by Hennlein et al., J Cell Biol. (2023) clearly showed that Smn deficiency impairs the F-actin dependent translocation of the high-affinity BDNF receptor TrkB to the cell surface resulting in reduced BDNF-mediated TrkB activation in motor axon terminals. Strikingly, the overexpression of PLS3 restores TrkB availability, and significantly improves the clustering of the active zone-associated voltage-gated calcium channel Ca<sub>v</sub>2.2 in growth cones of Smn-deficient motoneurons. These observations raise the question of how PLS3 mediates the proper cell surface localization and cluster-like formation of Ca<sub>v</sub>2.2 in motor axon terminals.</p>","PeriodicalId":36527,"journal":{"name":"Neuroscience Insights","volume":"19 ","pages":"26331055241226623"},"PeriodicalIF":2.6000,"publicationDate":"2024-01-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10799582/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neuroscience Insights","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/26331055241226623","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
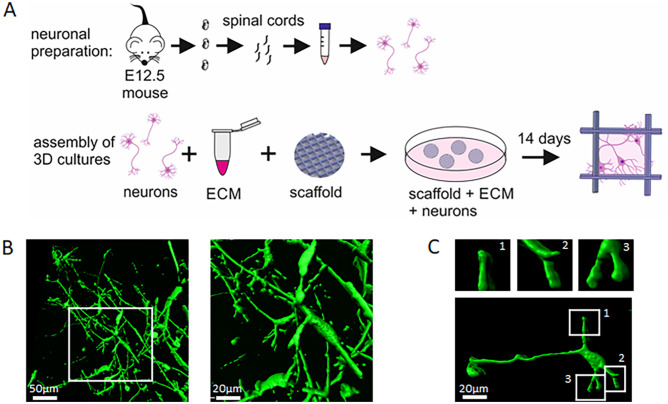
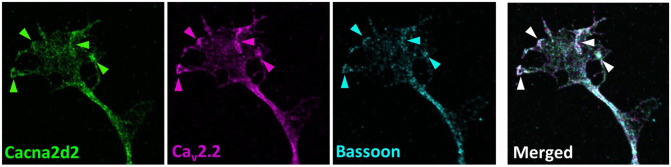
Loss of the Survival Motor Neuron (SMN) gene inevitably leads to spinal muscular atrophy (SMA), one of the most common fatal neuromuscular diseases in children with FDA and EMA approved therapies. However, the cellular mechanisms leading to neuromuscular junction (NMJ) dysfunction due to impaired Ca2+ homeostasis in the presynaptic compartment remain largely unexplained. In the last decade, the so-called SMA modifiers have gained attention. The F-actin bundler Plastin 3 (PLS3) is one of them and counteracts neurotransmission defects, including altered vesicle endocytosis, in Smn-deficient NMJs. Properly bundled F-actin is the basis for the translocation and arrangement of transmembrane proteins at the cell surface. Our recently published data by Hennlein et al., J Cell Biol. (2023) clearly showed that Smn deficiency impairs the F-actin dependent translocation of the high-affinity BDNF receptor TrkB to the cell surface resulting in reduced BDNF-mediated TrkB activation in motor axon terminals. Strikingly, the overexpression of PLS3 restores TrkB availability, and significantly improves the clustering of the active zone-associated voltage-gated calcium channel Cav2.2 in growth cones of Smn-deficient motoneurons. These observations raise the question of how PLS3 mediates the proper cell surface localization and cluster-like formation of Cav2.2 in motor axon terminals.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


