Gonench Kilich, Kelly Hassey, Edward M Behrens, Marni Falk, Adeline Vanderver, Daniel J Rader, Patrick J Cahill, Anna Raper, Zhe Zhang, Dawn Westerfer, Tanaya Jadhav, Laura Conlin, Kosuke Izumi, Ramakrishnan Rajagopalan, Kathleen E Sullivan
{"title":"Kagami Ogata syndrome: a small deletion refines critical region for imprinting.","authors":"Gonench Kilich, Kelly Hassey, Edward M Behrens, Marni Falk, Adeline Vanderver, Daniel J Rader, Patrick J Cahill, Anna Raper, Zhe Zhang, Dawn Westerfer, Tanaya Jadhav, Laura Conlin, Kosuke Izumi, Ramakrishnan Rajagopalan, Kathleen E Sullivan","doi":"10.1038/s41525-023-00389-2","DOIUrl":null,"url":null,"abstract":"<p><p>Kagami-Ogata syndrome is a rare imprinting disorder and its phenotypic overlap with multiple different etiologies hampers diagnosis. Genetic etiologies include paternal uniparental isodisomy (upd(14)pat), maternal allele deletions of differentially methylated regions (DMR) in 14q32.2 or pure primary epimutations. We report a patient with Kagami-Ogata syndrome and an atypical diagnostic odyssey with several negative standard-of-care genetic tests followed by epigenetic testing using methylation microarray and a targeted analysis of whole-genome sequencing to reveal a 203 bp deletion involving the MEG3 transcript and MEG3:TSS-DMR. Long-read sequencing enabled the simultaneous detection of the deletion, phasing, and biallelic hypermethylation of the MEG3:TSS-DMR region in a single assay. This case highlights the challenges in the sequential genetic testing paradigm, the utility of long-read sequencing as a single comprehensive diagnostic assay, and the smallest reported deletion causing Kagami-Ogata syndrome allowing important insights into the mechanism of imprinting effects at this locus.</p>","PeriodicalId":19273,"journal":{"name":"NPJ Genomic Medicine","volume":"9 1","pages":"5"},"PeriodicalIF":4.8000,"publicationDate":"2024-01-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10784583/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41525-023-00389-2","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
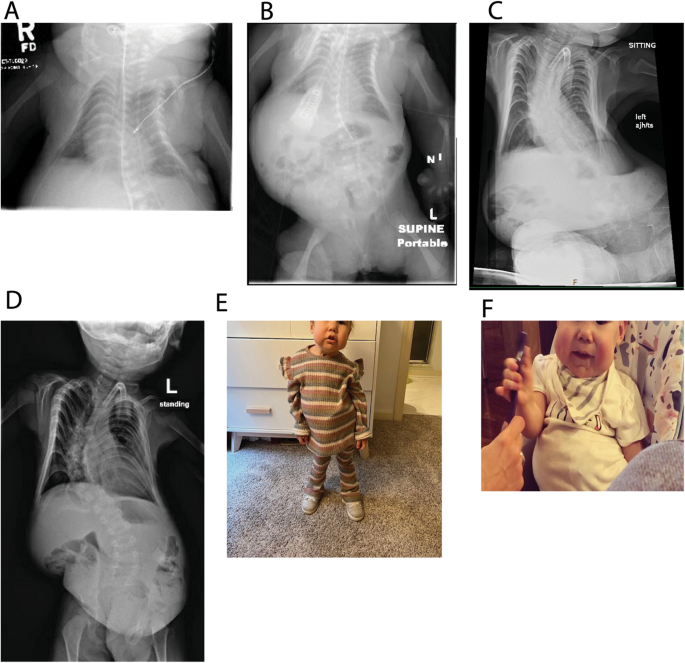
Kagami-Ogata syndrome is a rare imprinting disorder and its phenotypic overlap with multiple different etiologies hampers diagnosis. Genetic etiologies include paternal uniparental isodisomy (upd(14)pat), maternal allele deletions of differentially methylated regions (DMR) in 14q32.2 or pure primary epimutations. We report a patient with Kagami-Ogata syndrome and an atypical diagnostic odyssey with several negative standard-of-care genetic tests followed by epigenetic testing using methylation microarray and a targeted analysis of whole-genome sequencing to reveal a 203 bp deletion involving the MEG3 transcript and MEG3:TSS-DMR. Long-read sequencing enabled the simultaneous detection of the deletion, phasing, and biallelic hypermethylation of the MEG3:TSS-DMR region in a single assay. This case highlights the challenges in the sequential genetic testing paradigm, the utility of long-read sequencing as a single comprehensive diagnostic assay, and the smallest reported deletion causing Kagami-Ogata syndrome allowing important insights into the mechanism of imprinting effects at this locus.
NPJ Genomic MedicineBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
9.40
自引率
1.90%
发文量
67
审稿时长
17 weeks
期刊介绍:
npj Genomic Medicine is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.
The journal defines genomic medicine as "diagnosis, prognosis, prevention and/or treatment of disease and disorders of the mind and body, using approaches informed or enabled by knowledge of the genome and the molecules it encodes." Relevant and high-impact papers that encompass studies of individuals, families, or populations are considered for publication. An emphasis will include coupling detailed phenotype and genome sequencing information, both enabled by new technologies and informatics, to delineate the underlying aetiology of disease. Clinical recommendations and/or guidelines of how that data should be used in the clinical management of those patients in the study, and others, are also encouraged.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


