Yingsheng Zhang, Meng-Ju Wu, Wan-Chi Lu, Yi-Chuan Li, Chun Ju Chang, Jer-Yen Yang
{"title":"Metabolic switch regulates lineage plasticity and induces synthetic lethality in triple-negative breast cancer.","authors":"Yingsheng Zhang, Meng-Ju Wu, Wan-Chi Lu, Yi-Chuan Li, Chun Ju Chang, Jer-Yen Yang","doi":"10.1016/j.cmet.2023.12.003","DOIUrl":null,"url":null,"abstract":"<p><p>Metabolic reprogramming is key for cancer development, yet the mechanism that sustains triple-negative breast cancer (TNBC) cell growth despite deficient pyruvate kinase M2 (PKM2) and tumor glycolysis remains to be determined. Here, we find that deficiency in tumor glycolysis activates a metabolic switch from glycolysis to fatty acid β-oxidation (FAO) to fuel TNBC growth. We show that, in TNBC cells, PKM2 directly interacts with histone methyltransferase EZH2 to coordinately mediate epigenetic silencing of a carnitine transporter, SLC16A9. Inhibition of PKM2 leads to impaired EZH2 recruitment to SLC16A9, and in turn de-represses SLC16A9 expression to increase intracellular carnitine influx, programming TNBC cells to an FAO-dependent and luminal-like cell state. Together, these findings reveal a new metabolic switch that drives TNBC from a metabolically heterogeneous-lineage plastic cell state to an FAO-dependent-lineage committed cell state, where dual targeting of EZH2 and FAO induces potent synthetic lethality in TNBC.</p>","PeriodicalId":93927,"journal":{"name":"Cell metabolism","volume":"36 1","pages":"193-208.e8"},"PeriodicalIF":30.9000,"publicationDate":"2024-01-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell metabolism","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.cmet.2023.12.003","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
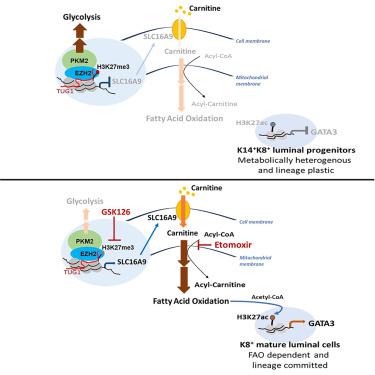
Metabolic reprogramming is key for cancer development, yet the mechanism that sustains triple-negative breast cancer (TNBC) cell growth despite deficient pyruvate kinase M2 (PKM2) and tumor glycolysis remains to be determined. Here, we find that deficiency in tumor glycolysis activates a metabolic switch from glycolysis to fatty acid β-oxidation (FAO) to fuel TNBC growth. We show that, in TNBC cells, PKM2 directly interacts with histone methyltransferase EZH2 to coordinately mediate epigenetic silencing of a carnitine transporter, SLC16A9. Inhibition of PKM2 leads to impaired EZH2 recruitment to SLC16A9, and in turn de-represses SLC16A9 expression to increase intracellular carnitine influx, programming TNBC cells to an FAO-dependent and luminal-like cell state. Together, these findings reveal a new metabolic switch that drives TNBC from a metabolically heterogeneous-lineage plastic cell state to an FAO-dependent-lineage committed cell state, where dual targeting of EZH2 and FAO induces potent synthetic lethality in TNBC.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


