Renjie Zhao, Ziyue Zou, John D. Weeks* and Pratyush Tiwary*,
{"title":"Quantifying the Relevance of Long-Range Forces for Crystal Nucleation in Water","authors":"Renjie Zhao, Ziyue Zou, John D. Weeks* and Pratyush Tiwary*, ","doi":"10.1021/acs.jctc.3c01120","DOIUrl":null,"url":null,"abstract":"<p >Understanding nucleation from aqueous solutions is of fundamental importance in a multitude of fields, ranging from materials science to biophysics. The complex solvent-mediated interactions in aqueous solutions hamper the development of a simple physical picture, elucidating the roles of different interactions in nucleation processes. In this work, we make use of three complementary techniques to disentangle the role played by short- and long-range interactions in solvent-mediated nucleation. Specifically, the first approach we utilize is the local molecular field (LMF) theory to renormalize long-range Coulomb electrostatics. Second, we use well-tempered metadynamics to speed up rare events governed by short-range interactions. Third, the deep learning-based State Predictive Information Bottleneck approach is employed in analyzing the reaction coordinate of the nucleation processes obtained from the LMF treatment coupled with well-tempered metadynamics. We find that the two-step nucleation mechanism can largely be captured by the short-range interactions, while the long-range interactions further contribute to the stability of the primary crystal state under ambient conditions. Furthermore, by analyzing the reaction coordinate obtained from the combined LMF-metadynamics treatment, we discern the fluctuations on different time scales, highlighting the need for long-range interactions when accounting for metastability.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"19 24","pages":"9093–9101"},"PeriodicalIF":5.5000,"publicationDate":"2023-12-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.3c01120","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
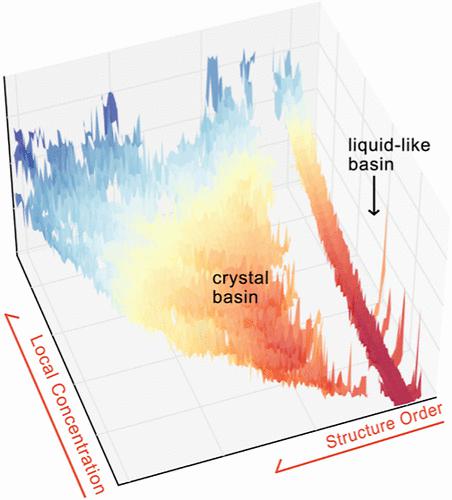
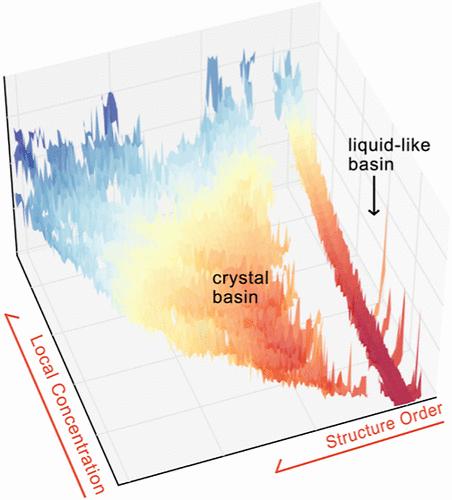
Understanding nucleation from aqueous solutions is of fundamental importance in a multitude of fields, ranging from materials science to biophysics. The complex solvent-mediated interactions in aqueous solutions hamper the development of a simple physical picture, elucidating the roles of different interactions in nucleation processes. In this work, we make use of three complementary techniques to disentangle the role played by short- and long-range interactions in solvent-mediated nucleation. Specifically, the first approach we utilize is the local molecular field (LMF) theory to renormalize long-range Coulomb electrostatics. Second, we use well-tempered metadynamics to speed up rare events governed by short-range interactions. Third, the deep learning-based State Predictive Information Bottleneck approach is employed in analyzing the reaction coordinate of the nucleation processes obtained from the LMF treatment coupled with well-tempered metadynamics. We find that the two-step nucleation mechanism can largely be captured by the short-range interactions, while the long-range interactions further contribute to the stability of the primary crystal state under ambient conditions. Furthermore, by analyzing the reaction coordinate obtained from the combined LMF-metadynamics treatment, we discern the fluctuations on different time scales, highlighting the need for long-range interactions when accounting for metastability.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


