Hi Jung Park, Eun Ah Choi, Sung Min Choi, Young-Ki Choi, Jae Il Lee, Kyeong Cheon Jung
{"title":"IL-4/IL-4 Ab complex enhances the accumulation of both antigen-specific and bystander CD8 T cells in mouse lungs infected with influenza A virus.","authors":"Hi Jung Park, Eun Ah Choi, Sung Min Choi, Young-Ki Choi, Jae Il Lee, Kyeong Cheon Jung","doi":"10.1186/s42826-023-00183-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Unlike conventional T cells, innate and virtual-memory CD8 T cells in naïve mice acquire their memory phenotypes and functions in the absence of antigenic encounters in a cytokine-dependent manner. The relevant cytokines include interleukin-4 (IL-4), type I interferon, and interleukin-15 (IL-15). Moreover, exogenous IL-4 can also induce de novo generation and/or expansion of the virtual-memory CD8 T cell population. In this study, we investigated whether exogenous IL-4 could enhance the immune response to a viral infection.</p><p><strong>Results: </strong>In vivo administration of IL-4 and an anti-IL-4 antibody complex (IL-4C) increased CXCR3 expression in both memory and naïve phenotype CD8 T cells in the absence of antigenic stimulation, and protected mice from lethal influenza infection. Flow cytometric analysis of lung-infiltrating immune cells on day 5 after virus infection revealed higher numbers of antigen-specific and bystander CD8 T cells in IL-4C-treated mice than in control mice. In particular, the bystander CD8 T cells were a naïve or evident memory phenotypes. Crucially, an anti-CXCR3 blocking antibody abrogated this IL-4C effect, reflecting that the increased accumulation of CD8 T cells in the lungs after IL-4C treatment is dependent on CXCR3.</p><p><strong>Conclusions: </strong>These data demonstrate that exogenous IL-4C plays a protective role by enhancing CXCR3-dependent migration of CD8 T cells into influenza-infected lungs.</p>","PeriodicalId":17993,"journal":{"name":"Laboratory Animal Research","volume":"39 1","pages":"32"},"PeriodicalIF":2.9000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10691054/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Laboratory Animal Research","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s42826-023-00183-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Unlike conventional T cells, innate and virtual-memory CD8 T cells in naïve mice acquire their memory phenotypes and functions in the absence of antigenic encounters in a cytokine-dependent manner. The relevant cytokines include interleukin-4 (IL-4), type I interferon, and interleukin-15 (IL-15). Moreover, exogenous IL-4 can also induce de novo generation and/or expansion of the virtual-memory CD8 T cell population. In this study, we investigated whether exogenous IL-4 could enhance the immune response to a viral infection.
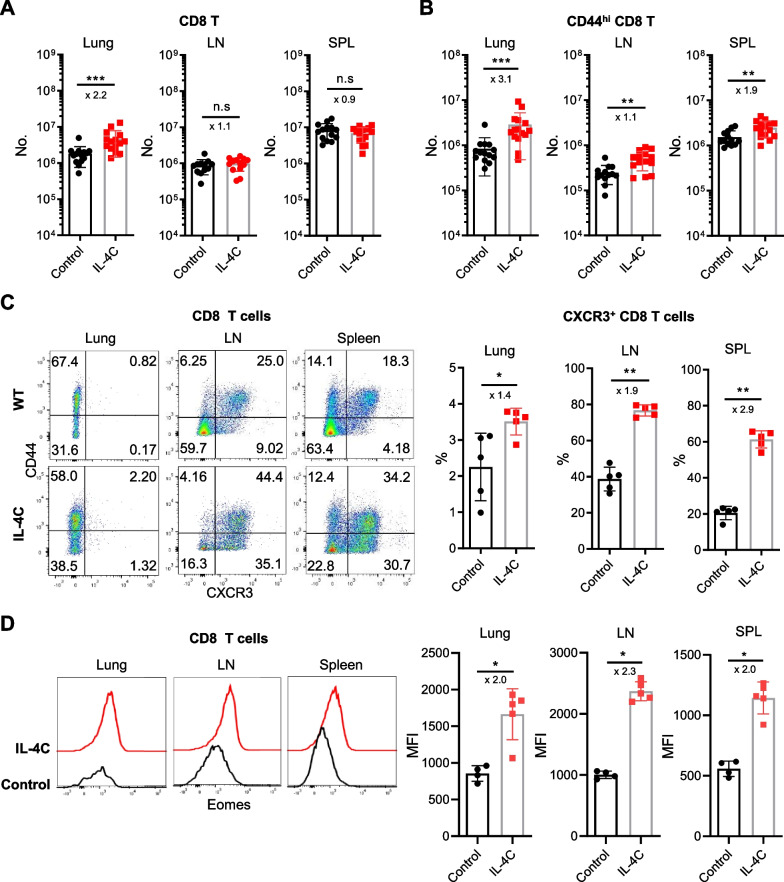
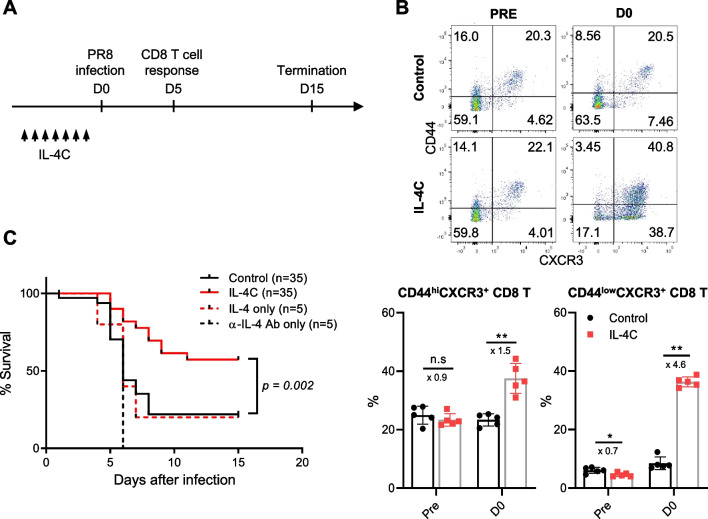
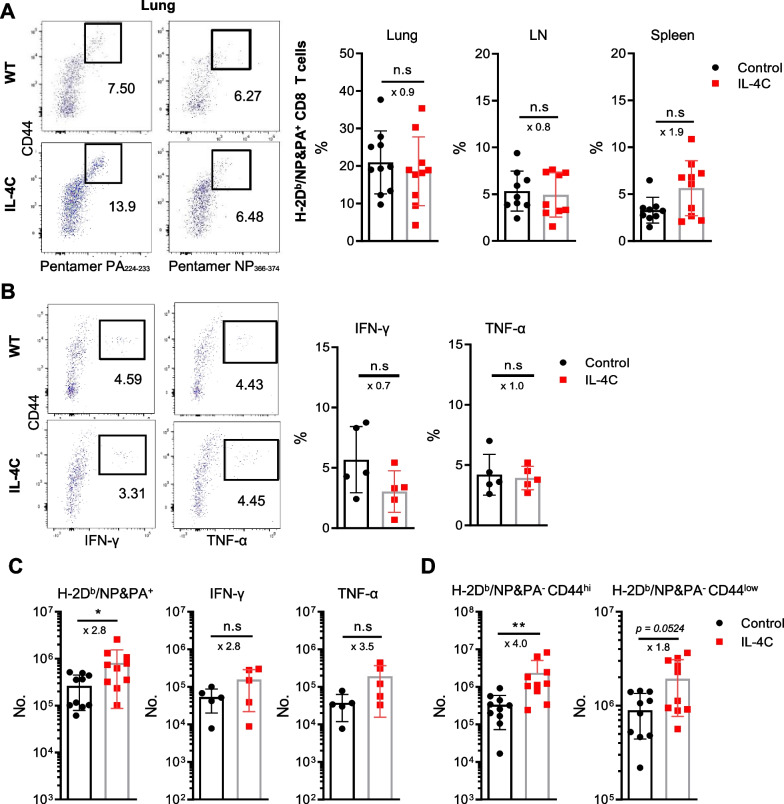
Results: In vivo administration of IL-4 and an anti-IL-4 antibody complex (IL-4C) increased CXCR3 expression in both memory and naïve phenotype CD8 T cells in the absence of antigenic stimulation, and protected mice from lethal influenza infection. Flow cytometric analysis of lung-infiltrating immune cells on day 5 after virus infection revealed higher numbers of antigen-specific and bystander CD8 T cells in IL-4C-treated mice than in control mice. In particular, the bystander CD8 T cells were a naïve or evident memory phenotypes. Crucially, an anti-CXCR3 blocking antibody abrogated this IL-4C effect, reflecting that the increased accumulation of CD8 T cells in the lungs after IL-4C treatment is dependent on CXCR3.
Conclusions: These data demonstrate that exogenous IL-4C plays a protective role by enhancing CXCR3-dependent migration of CD8 T cells into influenza-infected lungs.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


