A large single‐center cohort of bare lymphocyte syndrome: Immunological and genetic features in Turkey
IF 4.1
4区 医学
Q2 IMMUNOLOGY
引用次数: 0
Abstract
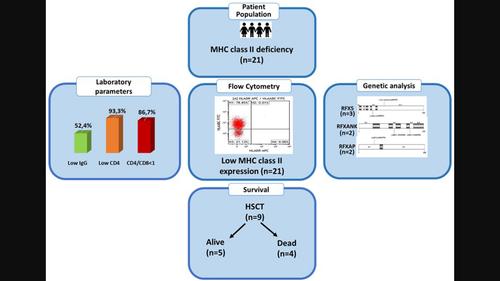
Abstract Major histocompatibility complex class II (MHC‐II) deficiency or bare lymphocyte syndrome (BLS) is a rare, early‐onset, autosomal recessive, and life‐threatening inborn error of immunity. We aimed to assess the demographic, clinical, laboratory, follow‐up, and treatment characteristics of patients with MHC‐II deficiency, together with their survival. We retrospectively investigated 21 patients with MHC‐II deficiency. Female/male ratio was 1.63. The median age at diagnosis was 16.3 months (5 months–9.7 years). Nineteen patients (90.5%) had parental consanguinity. Pulmonary diseases (pneumonia, chronic lung disease) (81%), diarrhoea (47.6%), and candidiasis (28.6%) were common. Four (19%) had autoimmunity, two developed septic arthritis, and three (14%) developed bronchiectasis in the follow‐up. Three patients (14%) had CMV viraemia, one with bilateral CMV retinitis. Eight (38.1%) had lymphocytopenia, and four (19%) had neutropenia. Serum IgM, IgA, and IgG levels were low in 18 (85.7%), 15 (71.4%), and 11 (52.4%) patients, respectively. CD4+ lymphocytopenia, a reversed CD4+/CD8+ ratio, and absent/low HLA‐DR expressions were detected in 93.3%, 86.7%, and 100% of the patients, respectively. Haematopoietic stem cell transplantation (HSCT) was performed on nine patients, and four died of septicaemia and ARDS after HSCT. The present median age of patients survived is 14 years (1–31 years). Genetic analysis was performed in 10 patients. RFX5 homozygous gene defect was found in three patients (P1, P4 and P8), and RFXANK (P2 and P14) and RFXAP (P18 and P19) heterozygous gene defects were found in each two patients, respectively. This large cohort showed that BLS patients have severe combined immunodeficiency (SCID)‐like clinical findings. Flow cytometric MHC‐II expression study is crucial for the diagnosis, differential diagnosis with SCID, early haematopoietic stem cell transplantation (HSCT), and post‐HSCT follow‐up. Genetic studies are required first for matched family donor evaluation before HSCT and then for genetic counselling.
裸淋巴细胞综合征的大型单中心队列研究:土耳其的免疫学和遗传学特征
主要组织相容性复合体II类(MHC‐II)缺乏或裸淋巴细胞综合征(BLS)是一种罕见的、早发的、常染色体隐性遗传的、危及生命的先天性免疫错误。我们的目的是评估MHC - II缺乏症患者的人口学、临床、实验室、随访和治疗特征,以及他们的生存率。我们回顾性调查了21例MHC‐II缺乏症患者。男女比例为1.63。诊断时的中位年龄为16.3个月(5个月- 9.7岁)。有亲本亲属19例(90.5%)。肺部疾病(肺炎、慢性肺病)(81%)、腹泻(47.6%)和念珠菌病(28.6%)最为常见。在随访中,4例(19%)患有自身免疫,2例发展为脓毒性关节炎,3例(14%)发展为支气管扩张。3例(14%)有巨细胞病毒血症,1例有双侧巨细胞病毒视网膜炎。淋巴细胞减少8例(38.1%),中性粒细胞减少4例(19%)。血清IgM、IgA和IgG水平低的患者分别为18例(85.7%)、15例(71.4%)和11例(52.4%)。CD4+淋巴细胞减少症、CD4+/CD8+比值逆转、HLA - DR表达缺失/低分别在93.3%、86.7%和100%的患者中检测到。9例患者行造血干细胞移植(HSCT), 4例患者在移植后死于败血症和ARDS。目前存活患者的中位年龄为14岁(1-31岁)。对10例患者进行遗传分析。3例患者(P1、P4、P8)存在RFX5纯合子基因缺陷,2例患者分别存在RFXANK (P2、P14)和RFXAP (P18、P19)杂合子基因缺陷。这一大型队列研究显示,BLS患者具有严重的联合免疫缺陷(SCID)样临床表现。流式细胞术MHC - II表达研究对于SCID的诊断、鉴别诊断、早期造血干细胞移植(HSCT)以及HSCT后随访至关重要。在造血干细胞移植前,首先需要进行遗传研究,以便对匹配的家庭供体进行评估,然后进行遗传咨询。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
7.70
自引率
5.40%
发文量
109
审稿时长
1 months
期刊介绍:
This peer-reviewed international journal publishes original articles and reviews on all aspects of basic, translational and clinical immunology. The journal aims to provide high quality service to authors, and high quality articles for readers.
The journal accepts for publication material from investigators all over the world, which makes a significant contribution to basic, translational and clinical immunology.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


