Areti-Maria Vasilogianni, Sarah Alrubia, Eman El-Khateeb, Zubida M. Al-Majdoub, Narciso Couto, Brahim Achour, Amin Rostami-Hodjegan and Jill Barber
{"title":"Complementarity of two proteomic data analysis tools in the identification of drug-metabolising enzymes and transporters in human liver†","authors":"Areti-Maria Vasilogianni, Sarah Alrubia, Eman El-Khateeb, Zubida M. Al-Majdoub, Narciso Couto, Brahim Achour, Amin Rostami-Hodjegan and Jill Barber","doi":"10.1039/D3MO00144J","DOIUrl":null,"url":null,"abstract":"<p >Several software packages are available for the analysis of proteomic LC-MS/MS data, including commercial (<em>e.g.</em> Mascot/Progenesis LC-MS) and open access software (<em>e.g.</em> MaxQuant). In this study, Progenesis and MaxQuant were used to analyse the same data set from human liver microsomes (<em>n</em> = 23). Comparison focussed on the total number of peptides and proteins identified by the two packages. For the peptides exclusively identified by each software package, distribution of peptide length, hydrophobicity, molecular weight, isoelectric point and score were compared. Using standard cut-off peptide scores, we found an average of only 65% overlap in detected peptides, with surprisingly little consistency in the characteristics of peptides exclusively detected by each package. Generally, MaxQuant detected more peptides than Progenesis, and the additional peptides were longer and had relatively lower scores. Progenesis-specific peptides tended to be more hydrophilic and basic relative to peptides detected only by MaxQuant. At the protein level, we focussed on drug-metabolising enzymes (DMEs) and transporters, by comparing the number of unique peptides detected by the two packages for these specific proteins of interest, and their abundance. The abundance of DMEs and SLC transporters showed good correlation between the two software tools, but ABC showed less consistency. In conclusion, in order to maximise the use of MS datasets, we recommend processing with more than one software package. Together, Progenesis and MaxQuant provided excellent coverage, with a core of common peptides identified in a very robust way.</p>","PeriodicalId":19065,"journal":{"name":"Molecular omics","volume":" 2","pages":" 115-127"},"PeriodicalIF":3.0000,"publicationDate":"2023-11-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/mo/d3mo00144j?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular omics","FirstCategoryId":"99","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/mo/d3mo00144j","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
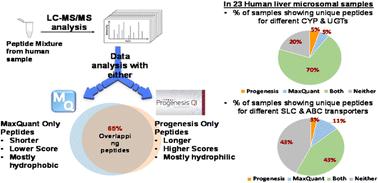
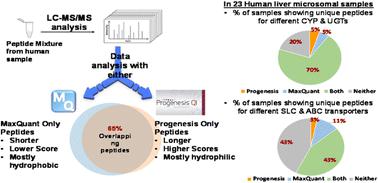
Several software packages are available for the analysis of proteomic LC-MS/MS data, including commercial (e.g. Mascot/Progenesis LC-MS) and open access software (e.g. MaxQuant). In this study, Progenesis and MaxQuant were used to analyse the same data set from human liver microsomes (n = 23). Comparison focussed on the total number of peptides and proteins identified by the two packages. For the peptides exclusively identified by each software package, distribution of peptide length, hydrophobicity, molecular weight, isoelectric point and score were compared. Using standard cut-off peptide scores, we found an average of only 65% overlap in detected peptides, with surprisingly little consistency in the characteristics of peptides exclusively detected by each package. Generally, MaxQuant detected more peptides than Progenesis, and the additional peptides were longer and had relatively lower scores. Progenesis-specific peptides tended to be more hydrophilic and basic relative to peptides detected only by MaxQuant. At the protein level, we focussed on drug-metabolising enzymes (DMEs) and transporters, by comparing the number of unique peptides detected by the two packages for these specific proteins of interest, and their abundance. The abundance of DMEs and SLC transporters showed good correlation between the two software tools, but ABC showed less consistency. In conclusion, in order to maximise the use of MS datasets, we recommend processing with more than one software package. Together, Progenesis and MaxQuant provided excellent coverage, with a core of common peptides identified in a very robust way.
Molecular omicsBiochemistry, Genetics and Molecular Biology-Biochemistry
CiteScore
5.40
自引率
3.40%
发文量
91
期刊介绍:
Molecular Omics publishes high-quality research from across the -omics sciences.
Topics include, but are not limited to:
-omics studies to gain mechanistic insight into biological processes – for example, determining the mode of action of a drug or the basis of a particular phenotype, such as drought tolerance
-omics studies for clinical applications with validation, such as finding biomarkers for diagnostics or potential new drug targets
-omics studies looking at the sub-cellular make-up of cells – for example, the subcellular localisation of certain proteins or post-translational modifications or new imaging techniques
-studies presenting new methods and tools to support omics studies, including new spectroscopic/chromatographic techniques, chip-based/array technologies and new classification/data analysis techniques. New methods should be proven and demonstrate an advance in the field.
Molecular Omics only accepts articles of high importance and interest that provide significant new insight into important chemical or biological problems. This could be fundamental research that significantly increases understanding or research that demonstrates clear functional benefits.
Papers reporting new results that could be routinely predicted, do not show a significant improvement over known research, or are of interest only to the specialist in the area are not suitable for publication in Molecular Omics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


