Tal Gilboa, Naama Elefant, Vardiella Meiner, Nuphar Hacohen
{"title":"Delineating the phenotype and genetic basis of AMPD2-related pontocerebellar hypoplasia.","authors":"Tal Gilboa, Naama Elefant, Vardiella Meiner, Nuphar Hacohen","doi":"10.1007/s10048-022-00706-4","DOIUrl":null,"url":null,"abstract":"<p><p>Pontocerebellar hypoplasia is a group of disorders with a wide range of presentations. We describe here the genetic and phenotypic features of PCH type 9 due to mutations in AMPD2. All patients have severe intellectual disability, and the vast majority manifest abnormal tone, cortical blindness, and microcephaly. Almost all have agenesis of the corpus callosum and severe cerebellar hypoplasia. The course is not progressive, however, few die in the first decade of life. Mutations are spread throughout the gene, and no hot spot can be identified. One of the mutations we report here is the most distal truncating variant known in this gene and is predicted to result in a truncated protein. The phenotype is severe in all cases; thus, no clear genotype-phenotype correlation can be established.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"24 1","pages":"61-66"},"PeriodicalIF":1.6000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-022-00706-4","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
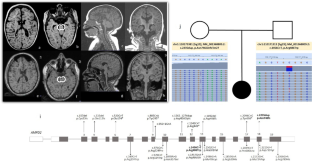
Pontocerebellar hypoplasia is a group of disorders with a wide range of presentations. We describe here the genetic and phenotypic features of PCH type 9 due to mutations in AMPD2. All patients have severe intellectual disability, and the vast majority manifest abnormal tone, cortical blindness, and microcephaly. Almost all have agenesis of the corpus callosum and severe cerebellar hypoplasia. The course is not progressive, however, few die in the first decade of life. Mutations are spread throughout the gene, and no hot spot can be identified. One of the mutations we report here is the most distal truncating variant known in this gene and is predicted to result in a truncated protein. The phenotype is severe in all cases; thus, no clear genotype-phenotype correlation can be established.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


