C. Gary Olds, Jessie W. Berta-Thompson, Justin J. Loucks, Richard A. Levy, Andrew W. Wilson
{"title":"Applying a modified metabarcoding approach for the sequencing of macrofungal specimens from fungarium collections","authors":"C. Gary Olds, Jessie W. Berta-Thompson, Justin J. Loucks, Richard A. Levy, Andrew W. Wilson","doi":"10.1002/aps3.11508","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Premise</h3>\n \n <p>Fungaria are an underutilized resource for understanding fungal biodiversity. The effort and cost of producing DNA barcode sequence data for large numbers of fungal specimens can be prohibitive. This study applies a modified metabarcoding approach that provides a labor-efficient and cost-effective solution for sequencing the fungal DNA barcodes of hundreds of specimens at once.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>We applied a two-step PCR approach using nested, barcoded primers to sequence the fungal nrITS2 region of 766 macrofungal specimens using the Illumina platform. The specimens represent a broad taxonomic sampling of the Dikarya. Of these, 382 <i>Lactarius</i> specimens were analyzed to identify molecular operational taxonomic units (MOTUs) using a phylogenetic approach. The raw sequences were trimmed, filtered, assessed, and analyzed using the DADA2 amplicon de-noising toolkit and Biopython. The sequences were compared to the NCBI and UNITE databases and Sanger nrITS sequences from the same specimens.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>The taxonomic identities derived from the nrITS2 sequence data were >90% accurate across all specimens sampled. A phylogenetic analysis of the <i>Lactarius</i> sequences identified 20 MOTUs.</p>\n </section>\n \n <section>\n \n <h3> Discussion</h3>\n \n <p>The results demonstrate the capacity of these methods to produce nrITS2 sequences from large numbers of fungarium specimens. This provides an opportunity to more effectively use fungarium collections to advance fungal diversity identification and documentation.</p>\n </section>\n </div>","PeriodicalId":8022,"journal":{"name":"Applications in Plant Sciences","volume":"11 1","pages":""},"PeriodicalIF":2.7000,"publicationDate":"2023-02-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://bsapubs.onlinelibrary.wiley.com/doi/epdf/10.1002/aps3.11508","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Applications in Plant Sciences","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/aps3.11508","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"PLANT SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
Premise
Fungaria are an underutilized resource for understanding fungal biodiversity. The effort and cost of producing DNA barcode sequence data for large numbers of fungal specimens can be prohibitive. This study applies a modified metabarcoding approach that provides a labor-efficient and cost-effective solution for sequencing the fungal DNA barcodes of hundreds of specimens at once.
Methods
We applied a two-step PCR approach using nested, barcoded primers to sequence the fungal nrITS2 region of 766 macrofungal specimens using the Illumina platform. The specimens represent a broad taxonomic sampling of the Dikarya. Of these, 382 Lactarius specimens were analyzed to identify molecular operational taxonomic units (MOTUs) using a phylogenetic approach. The raw sequences were trimmed, filtered, assessed, and analyzed using the DADA2 amplicon de-noising toolkit and Biopython. The sequences were compared to the NCBI and UNITE databases and Sanger nrITS sequences from the same specimens.
Results
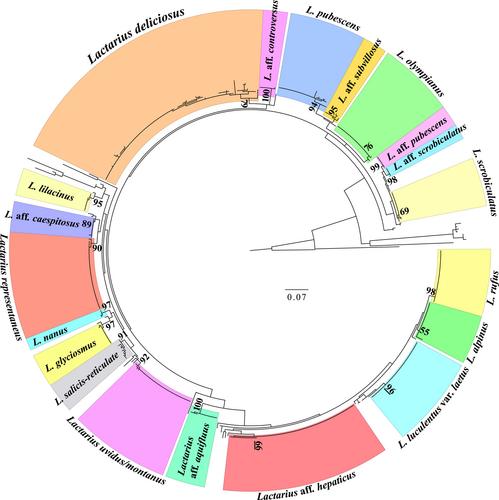
The taxonomic identities derived from the nrITS2 sequence data were >90% accurate across all specimens sampled. A phylogenetic analysis of the Lactarius sequences identified 20 MOTUs.
Discussion
The results demonstrate the capacity of these methods to produce nrITS2 sequences from large numbers of fungarium specimens. This provides an opportunity to more effectively use fungarium collections to advance fungal diversity identification and documentation.
期刊介绍:
Applications in Plant Sciences (APPS) is a monthly, peer-reviewed, open access journal promoting the rapid dissemination of newly developed, innovative tools and protocols in all areas of the plant sciences, including genetics, structure, function, development, evolution, systematics, and ecology. Given the rapid progress today in technology and its application in the plant sciences, the goal of APPS is to foster communication within the plant science community to advance scientific research. APPS is a publication of the Botanical Society of America, originating in 2009 as the American Journal of Botany''s online-only section, AJB Primer Notes & Protocols in the Plant Sciences.
APPS publishes the following types of articles: (1) Protocol Notes describe new methods and technological advancements; (2) Genomic Resources Articles characterize the development and demonstrate the usefulness of newly developed genomic resources, including transcriptomes; (3) Software Notes detail new software applications; (4) Application Articles illustrate the application of a new protocol, method, or software application within the context of a larger study; (5) Review Articles evaluate available techniques, methods, or protocols; (6) Primer Notes report novel genetic markers with evidence of wide applicability.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


