Svetlana Kocheva, Marija Gjorgijevska, Marija Vujovic, Kata Martinova, Zorica Antevska-Trajkova, Aleksandra Jovanovska, Katarina Stavrikj, Dijana Plaseska-Karanfilska
{"title":"Autoimmune lymphoproliferative syndrome identified through reverse phenotyping.","authors":"Svetlana Kocheva, Marija Gjorgijevska, Marija Vujovic, Kata Martinova, Zorica Antevska-Trajkova, Aleksandra Jovanovska, Katarina Stavrikj, Dijana Plaseska-Karanfilska","doi":"10.5114/ceji.2022.118079","DOIUrl":null,"url":null,"abstract":"<p><p>Autoimmune lymphoproliferative syndrome (ALPS) is a chronic non-malignant lymphoproliferative disorder caused by mutations in the genes involved in programmed cell death. It is inherited as an autosomal dominant pattern with variable penetrance. In this paper we present the first report of a Macedonian family with ALPS, caused by a novel heterozygous variant in the FAS gene. The next generation sequencing (NGS) analysis in a patient with splenomegaly, suspected for hereditary spherocytosis, showed presence of the FAS c.913dupA, p.Thr305AsnfsTer16 variant. The same variant was present in the patient's mother, but not in the mother's parents (proband's grandparents). Thus, the pathogenic FAS variant has arisen as a de novo event in the proband's mother. Later, analysis of the newborn affected sister showed presence of the same FAS variant. Additional clinical and laboratory investigations in the proband and her sister confirmed the presence of specific biomarkers for ALPS. A first-line NGS analysis allows identification of the genetic defect and initiation of appropriate clinical examinations to promptly establish the clinical diagnosis in patients with rare diseases. Reverse phenotyping in our case provided a prompt and accurate diagnosis and early initiation of specific therapy.</p>","PeriodicalId":9694,"journal":{"name":"Central European Journal of Immunology","volume":"47 2","pages":"179-182"},"PeriodicalIF":1.6000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/3b/52/CEJI-47-47481.PMC9894091.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Central European Journal of Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.5114/ceji.2022.118079","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
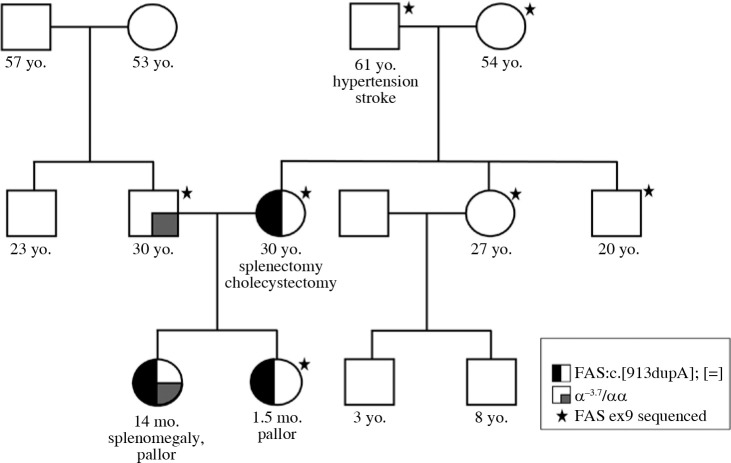
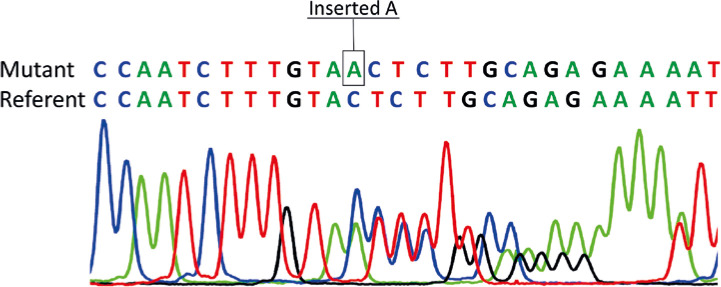
Autoimmune lymphoproliferative syndrome (ALPS) is a chronic non-malignant lymphoproliferative disorder caused by mutations in the genes involved in programmed cell death. It is inherited as an autosomal dominant pattern with variable penetrance. In this paper we present the first report of a Macedonian family with ALPS, caused by a novel heterozygous variant in the FAS gene. The next generation sequencing (NGS) analysis in a patient with splenomegaly, suspected for hereditary spherocytosis, showed presence of the FAS c.913dupA, p.Thr305AsnfsTer16 variant. The same variant was present in the patient's mother, but not in the mother's parents (proband's grandparents). Thus, the pathogenic FAS variant has arisen as a de novo event in the proband's mother. Later, analysis of the newborn affected sister showed presence of the same FAS variant. Additional clinical and laboratory investigations in the proband and her sister confirmed the presence of specific biomarkers for ALPS. A first-line NGS analysis allows identification of the genetic defect and initiation of appropriate clinical examinations to promptly establish the clinical diagnosis in patients with rare diseases. Reverse phenotyping in our case provided a prompt and accurate diagnosis and early initiation of specific therapy.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


