Raphaël Leman, Béatrice Parfait, Dominique Vidaud, Emmanuelle Girodon, Laurence Pacot, Gérald Le Gac, Chandran Ka, Claude Ferec, Yann Fichou, Céline Quesnelle, Camille Aucouturier, Etienne Muller, Dominique Vaur, Laurent Castera, Flavie Boulouard, Agathe Ricou, Hélène Tubeuf, Omar Soukarieh, Pascaline Gaildrat, Florence Riant, Marine Guillaud-Bataille, Sandrine M. Caputo, Virginie Caux-Moncoutier, Nadia Boutry-Kryza, Françoise Bonnet-Dorion, Ines Schultz, Maria Rossing, Olivier Quenez, Louis Goldenberg, Valentin Harter, Michael T. Parsons, Amanda B. Spurdle, Thierry Frébourg, Alexandra Martins, Claude Houdayer, Sophie Krieger
{"title":"SPiP: Splicing Prediction Pipeline, a machine learning tool for massive detection of exonic and intronic variant effects on mRNA splicing","authors":"Raphaël Leman, Béatrice Parfait, Dominique Vidaud, Emmanuelle Girodon, Laurence Pacot, Gérald Le Gac, Chandran Ka, Claude Ferec, Yann Fichou, Céline Quesnelle, Camille Aucouturier, Etienne Muller, Dominique Vaur, Laurent Castera, Flavie Boulouard, Agathe Ricou, Hélène Tubeuf, Omar Soukarieh, Pascaline Gaildrat, Florence Riant, Marine Guillaud-Bataille, Sandrine M. Caputo, Virginie Caux-Moncoutier, Nadia Boutry-Kryza, Françoise Bonnet-Dorion, Ines Schultz, Maria Rossing, Olivier Quenez, Louis Goldenberg, Valentin Harter, Michael T. Parsons, Amanda B. Spurdle, Thierry Frébourg, Alexandra Martins, Claude Houdayer, Sophie Krieger","doi":"10.1002/humu.24491","DOIUrl":null,"url":null,"abstract":"<p>Modeling splicing is essential for tackling the challenge of variant interpretation as each nucleotide variation can be pathogenic by affecting pre-mRNA splicing via disruption/creation of splicing motifs such as 5′/3′ splice sites, branch sites, or splicing regulatory elements. Unfortunately, most in silico tools focus on a specific type of splicing motif, which is why we developed the Splicing Prediction Pipeline (SPiP) to perform, in one single bioinformatic analysis based on a machine learning approach, a comprehensive assessment of the variant effect on different splicing motifs. We gathered a curated set of 4616 variants scattered all along the sequence of 227 genes, with their corresponding splicing studies. The Bayesian analysis provided us with the number of control variants, that is, variants without impact on splicing, to mimic the deluge of variants from high-throughput sequencing data. Results show that SPiP can deal with the diversity of splicing alterations, with 83.13% sensitivity and 99% specificity to detect spliceogenic variants. Overall performance as measured by area under the receiving operator curve was 0.986, better than SpliceAI and SQUIRLS (0.965 and 0.766) for the same data set. SPiP lends itself to a unique suite for comprehensive prediction of spliceogenicity in the genomic medicine era. SPiP is available at: https://sourceforge.net/projects/splicing-prediction-pipeline/</p>","PeriodicalId":13061,"journal":{"name":"Human Mutation","volume":"43 12","pages":"2308-2323"},"PeriodicalIF":3.7000,"publicationDate":"2022-10-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/humu.24491","citationCount":"17","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Mutation","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/humu.24491","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 17
Abstract
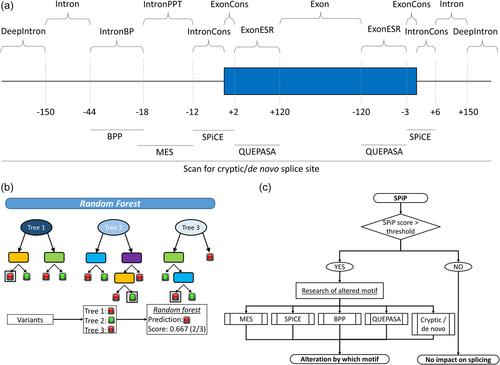
Modeling splicing is essential for tackling the challenge of variant interpretation as each nucleotide variation can be pathogenic by affecting pre-mRNA splicing via disruption/creation of splicing motifs such as 5′/3′ splice sites, branch sites, or splicing regulatory elements. Unfortunately, most in silico tools focus on a specific type of splicing motif, which is why we developed the Splicing Prediction Pipeline (SPiP) to perform, in one single bioinformatic analysis based on a machine learning approach, a comprehensive assessment of the variant effect on different splicing motifs. We gathered a curated set of 4616 variants scattered all along the sequence of 227 genes, with their corresponding splicing studies. The Bayesian analysis provided us with the number of control variants, that is, variants without impact on splicing, to mimic the deluge of variants from high-throughput sequencing data. Results show that SPiP can deal with the diversity of splicing alterations, with 83.13% sensitivity and 99% specificity to detect spliceogenic variants. Overall performance as measured by area under the receiving operator curve was 0.986, better than SpliceAI and SQUIRLS (0.965 and 0.766) for the same data set. SPiP lends itself to a unique suite for comprehensive prediction of spliceogenicity in the genomic medicine era. SPiP is available at: https://sourceforge.net/projects/splicing-prediction-pipeline/
期刊介绍:
Human Mutation is a peer-reviewed journal that offers publication of original Research Articles, Methods, Mutation Updates, Reviews, Database Articles, Rapid Communications, and Letters on broad aspects of mutation research in humans. Reports of novel DNA variations and their phenotypic consequences, reports of SNPs demonstrated as valuable for genomic analysis, descriptions of new molecular detection methods, and novel approaches to clinical diagnosis are welcomed. Novel reports of gene organization at the genomic level, reported in the context of mutation investigation, may be considered. The journal provides a unique forum for the exchange of ideas, methods, and applications of interest to molecular, human, and medical geneticists in academic, industrial, and clinical research settings worldwide.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


