{"title":"Metabolomics analysis of mycelial exudates provides insights into fungal antagonists of <i>Armillaria</i>.","authors":"Jian Zhan, Jing Yuan, Jianwei Liu, Fengming Zhang, Fuqiang Yu, Yanliang Wang","doi":"10.1080/21501203.2023.2238753","DOIUrl":null,"url":null,"abstract":"<p><p>The genus <i>Armillaria</i> has high edible and medical values, with zones of antagonism often occurring when different species are paired in culture on agar media, while the antagonism-induced metabolic alteration remains unclear. Here, the metabolome of mycelial exudates of two Chinese <i>Armillaria</i> biological species, C and G, co-cultured or cultured separately was analysed to discover the candidate biomarkers and the key metabolic pathways involved in <i>Armillaria</i> antagonists. A total of 2,377 metabolites were identified, mainly organic acids and derivatives, lipids and lipid-like molecules, and organoheterocyclic compounds. There were 248 and 142 differentially expressed metabolites between group C-G and C, C-G, and G, respectively, and fourteen common differentially expressed metabolites including malate, uracil, Leu-Gln-Arg, etc. Metabolic pathways like TCA cycle and pyrimidine metabolism were significantly affected by C-G co-culture. Additionally, 156 new metabolites (largely organic acids and derivatives) including 32 potential antifungal compounds, primarily enriched into biosynthesis of secondary metabolites pathways were identified in C-G co-culture mode. We concluded that malate and uracil could be used as the candidate biomarkers, and TCA cycle and pyrimidine metabolism were the key metabolic pathways involved in <i>Armillaria</i> antagonists. The metabolic changes revealed in this study provide insights into the mechanisms underlying fungal antagonists.</p>","PeriodicalId":18833,"journal":{"name":"Mycology","volume":"14 3","pages":"264-274"},"PeriodicalIF":4.4000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/02/ff/TMYC_14_2238753.PMC10424624.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Mycology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1080/21501203.2023.2238753","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MYCOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
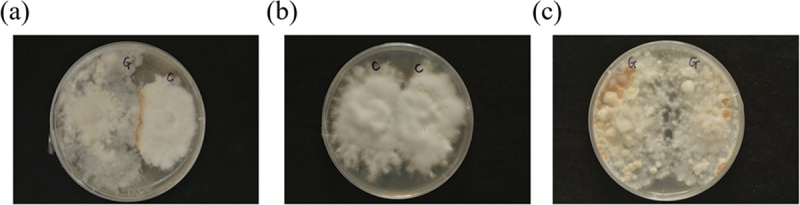
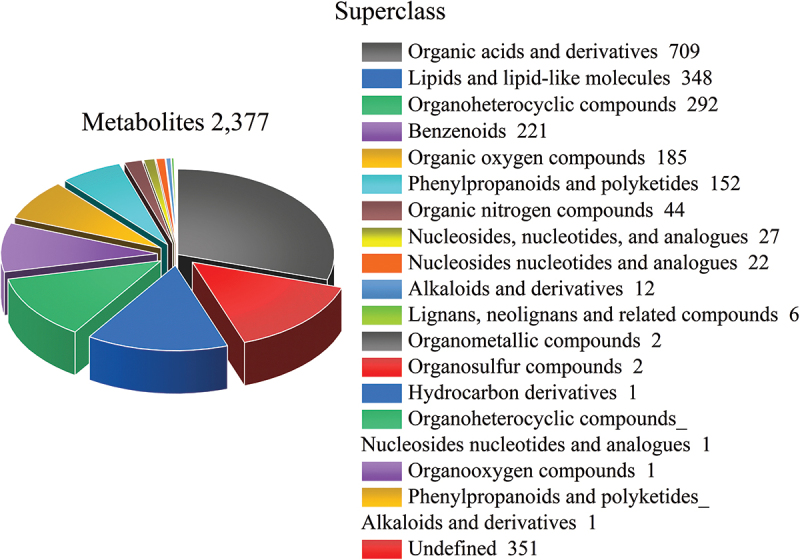
The genus Armillaria has high edible and medical values, with zones of antagonism often occurring when different species are paired in culture on agar media, while the antagonism-induced metabolic alteration remains unclear. Here, the metabolome of mycelial exudates of two Chinese Armillaria biological species, C and G, co-cultured or cultured separately was analysed to discover the candidate biomarkers and the key metabolic pathways involved in Armillaria antagonists. A total of 2,377 metabolites were identified, mainly organic acids and derivatives, lipids and lipid-like molecules, and organoheterocyclic compounds. There were 248 and 142 differentially expressed metabolites between group C-G and C, C-G, and G, respectively, and fourteen common differentially expressed metabolites including malate, uracil, Leu-Gln-Arg, etc. Metabolic pathways like TCA cycle and pyrimidine metabolism were significantly affected by C-G co-culture. Additionally, 156 new metabolites (largely organic acids and derivatives) including 32 potential antifungal compounds, primarily enriched into biosynthesis of secondary metabolites pathways were identified in C-G co-culture mode. We concluded that malate and uracil could be used as the candidate biomarkers, and TCA cycle and pyrimidine metabolism were the key metabolic pathways involved in Armillaria antagonists. The metabolic changes revealed in this study provide insights into the mechanisms underlying fungal antagonists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


