Zoe White, Zeren Sun, Elodie Sauge, Dan Cox, Graham Donen, Dmitri Pechkovsky, Volker Straub, Gordon A Francis, Pascal Bernatchez
{"title":"Limb-girdle muscular dystrophy type 2B causes HDL-C abnormalities in patients and statin-resistant muscle wasting in dysferlin-deficient mice.","authors":"Zoe White, Zeren Sun, Elodie Sauge, Dan Cox, Graham Donen, Dmitri Pechkovsky, Volker Straub, Gordon A Francis, Pascal Bernatchez","doi":"10.1186/s13395-022-00308-6","DOIUrl":null,"url":null,"abstract":"<p><p>Limb-girdle muscular dystrophy (MD) type 2B (LGMD2B) and Duchenne MD (DMD) are caused by mutations to the Dysferlin and Dystrophin genes, respectively. We have recently demonstrated in typically mild dysferlin- and dystrophin-deficient mouse models that increased plasma cholesterol levels severely exacerbate muscle wasting, and that DMD patients display primary dyslipidemia characterized by elevated plasma cholesterol and triglycerides. Herein, we investigate lipoprotein abnormalities in LGMD2B and if statin therapy protects dysferlin-deficient mice (Dysf) from muscle damage. Herein, lipoproteins and liver enzymes from LGMD2B patients and dysferlin-null (Dysf) mice were analyzed. Simvastatin, which exhibits anti-muscle wasting effects in mouse models of DMD and corrects aberrant expression of key markers of lipid metabolism and endogenous cholesterol synthesis, was tested in Dysf mice. Muscle damage and fibrosis were assessed by immunohistochemistry and cholesterol signalling pathways via Western blot. LGMD2B patients show reduced serum high-density lipoprotein cholesterol (HDL-C) levels compared to healthy controls and exhibit a greater prevalence of abnormal total cholesterol (CHOL)/HDL-C ratios despite an absence of liver dysfunction. While Dysf mice presented with reduced CHOL and associated HDL-C and LDL-C-associated fractions, simvastatin treatment did not prevent muscle wasting in quadriceps and triceps muscle groups or correct aberrant low-density lipoprotein receptor (LDLR) and 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) protein expression. LGMD2B patients present with reduced serum concentrations of HDL-C, a major metabolic comorbidity, and as a result, statin therapy is unlikely to prevent muscle wasting in this population. We propose that like DMD, LGMD2B should be considered as a new type of genetic dyslipidemia.</p>","PeriodicalId":21747,"journal":{"name":"Skeletal Muscle","volume":"12 1","pages":"25"},"PeriodicalIF":5.3000,"publicationDate":"2022-11-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9706908/pdf/","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Skeletal Muscle","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13395-022-00308-6","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 3
Abstract
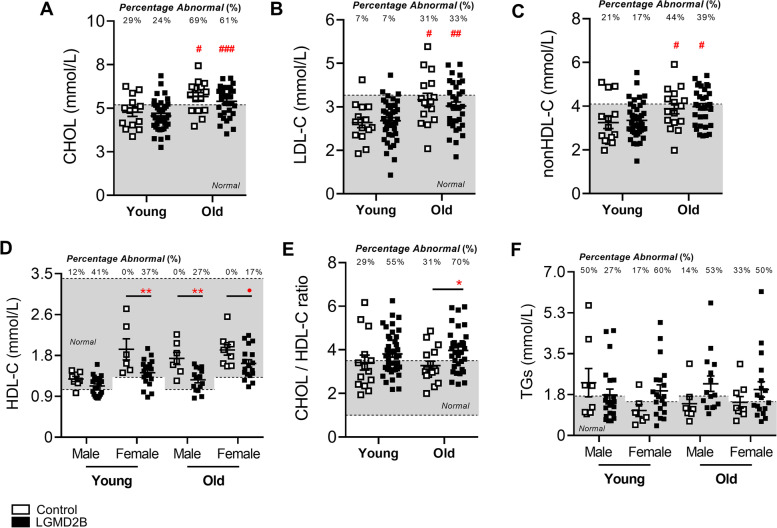
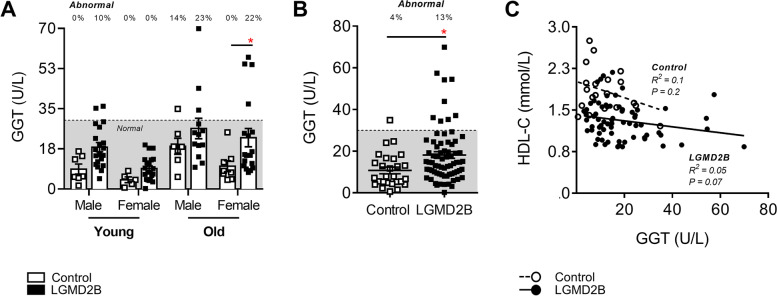
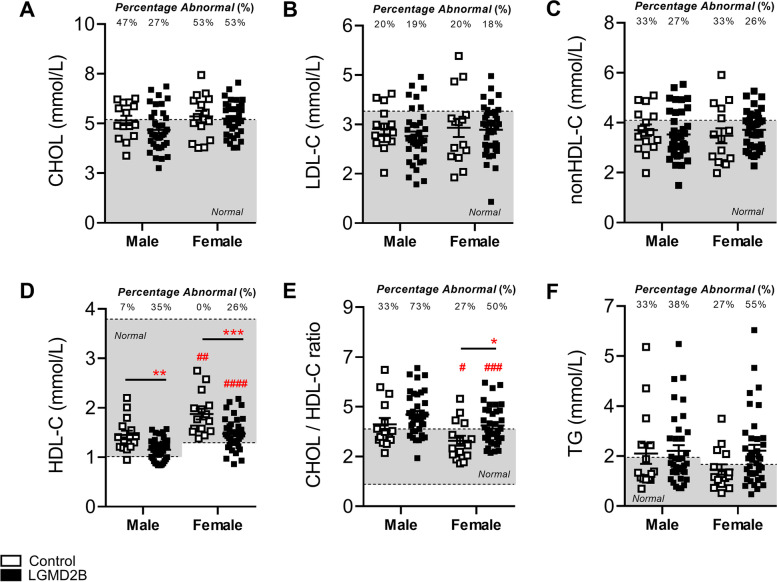
Limb-girdle muscular dystrophy (MD) type 2B (LGMD2B) and Duchenne MD (DMD) are caused by mutations to the Dysferlin and Dystrophin genes, respectively. We have recently demonstrated in typically mild dysferlin- and dystrophin-deficient mouse models that increased plasma cholesterol levels severely exacerbate muscle wasting, and that DMD patients display primary dyslipidemia characterized by elevated plasma cholesterol and triglycerides. Herein, we investigate lipoprotein abnormalities in LGMD2B and if statin therapy protects dysferlin-deficient mice (Dysf) from muscle damage. Herein, lipoproteins and liver enzymes from LGMD2B patients and dysferlin-null (Dysf) mice were analyzed. Simvastatin, which exhibits anti-muscle wasting effects in mouse models of DMD and corrects aberrant expression of key markers of lipid metabolism and endogenous cholesterol synthesis, was tested in Dysf mice. Muscle damage and fibrosis were assessed by immunohistochemistry and cholesterol signalling pathways via Western blot. LGMD2B patients show reduced serum high-density lipoprotein cholesterol (HDL-C) levels compared to healthy controls and exhibit a greater prevalence of abnormal total cholesterol (CHOL)/HDL-C ratios despite an absence of liver dysfunction. While Dysf mice presented with reduced CHOL and associated HDL-C and LDL-C-associated fractions, simvastatin treatment did not prevent muscle wasting in quadriceps and triceps muscle groups or correct aberrant low-density lipoprotein receptor (LDLR) and 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) protein expression. LGMD2B patients present with reduced serum concentrations of HDL-C, a major metabolic comorbidity, and as a result, statin therapy is unlikely to prevent muscle wasting in this population. We propose that like DMD, LGMD2B should be considered as a new type of genetic dyslipidemia.
期刊介绍:
The only open access journal in its field, Skeletal Muscle publishes novel, cutting-edge research and technological advancements that investigate the molecular mechanisms underlying the biology of skeletal muscle. Reflecting the breadth of research in this area, the journal welcomes manuscripts about the development, metabolism, the regulation of mass and function, aging, degeneration, dystrophy and regeneration of skeletal muscle, with an emphasis on understanding adult skeletal muscle, its maintenance, and its interactions with non-muscle cell types and regulatory modulators.
Main areas of interest include:
-differentiation of skeletal muscle-
atrophy and hypertrophy of skeletal muscle-
aging of skeletal muscle-
regeneration and degeneration of skeletal muscle-
biology of satellite and satellite-like cells-
dystrophic degeneration of skeletal muscle-
energy and glucose homeostasis in skeletal muscle-
non-dystrophic genetic diseases of skeletal muscle, such as Spinal Muscular Atrophy and myopathies-
maintenance of neuromuscular junctions-
roles of ryanodine receptors and calcium signaling in skeletal muscle-
roles of nuclear receptors in skeletal muscle-
roles of GPCRs and GPCR signaling in skeletal muscle-
other relevant aspects of skeletal muscle biology.
In addition, articles on translational clinical studies that address molecular and cellular mechanisms of skeletal muscle will be published. Case reports are also encouraged for submission.
Skeletal Muscle reflects the breadth of research on skeletal muscle and bridges gaps between diverse areas of science for example cardiac cell biology and neurobiology, which share common features with respect to cell differentiation, excitatory membranes, cell-cell communication, and maintenance. Suitable articles are model and mechanism-driven, and apply statistical principles where appropriate; purely descriptive studies are of lesser interest.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


