Saar Anis, Tsvia Fay-Karmon, Simon Lassman, Fadi Shbat, Orit Lesman-Segev, Nofar Mor, Ortal Barel, Dan Dominissini, Odelia Chorin, Elon Pras, Lior Greenbaum, Sharon Hassin-Baer
{"title":"Adult-onset Alexander disease among patients of Jewish Syrian descent.","authors":"Saar Anis, Tsvia Fay-Karmon, Simon Lassman, Fadi Shbat, Orit Lesman-Segev, Nofar Mor, Ortal Barel, Dan Dominissini, Odelia Chorin, Elon Pras, Lior Greenbaum, Sharon Hassin-Baer","doi":"10.1007/s10048-023-00732-w","DOIUrl":null,"url":null,"abstract":"<p><p>Alexander disease (AxD) is a rare autosomal dominant leukodystrophy caused by heterozygous mutations in the glial fibrillary acid protein (GFAP) gene. The age of symptoms onset ranges from infancy to adulthood, with variable clinical and radiological manifestations. Adult-onset AxD manifests as a chronic and progressive condition, characterized by bulbar, motor, cerebellar, and other clinical signs and symptoms. Neuroradiological findings typically involve the brainstem and cervical spinal cord. Adult-onset AxD has been described in diverse populations but is rare in Israel. We present a series of patients diagnosed with adult-onset AxD from three families, all of Jewish Syrian descent. Five patients (4 females) were diagnosed with adult-onset AxD due to the heterozygous mutation c.219G > A, p.Met73Ile in GFAP. Age at symptoms onset ranged from 48 to 61 years. Clinical characteristics were typical and involved progressive bulbar and gait disturbance, followed by pyramidal and cerebellar impairment, dysautonomia, and cognitive decline. Imaging findings included medullary and cervical spinal atrophy and mostly infratentorial white matter hyperintensities. A newly recognized cluster of adult-onset AxD in Jews of Syrian origin is presented. This disorder should be considered in differential diagnosis in appropriate circumstances. Genetic counselling for family members is required in order to discuss options for future family planning.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":" ","pages":"303-310"},"PeriodicalIF":1.6000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00732-w","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/2 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
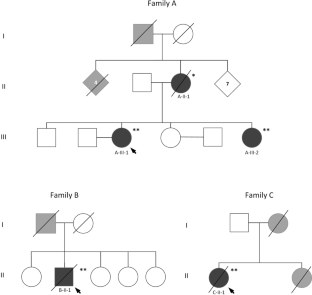
Alexander disease (AxD) is a rare autosomal dominant leukodystrophy caused by heterozygous mutations in the glial fibrillary acid protein (GFAP) gene. The age of symptoms onset ranges from infancy to adulthood, with variable clinical and radiological manifestations. Adult-onset AxD manifests as a chronic and progressive condition, characterized by bulbar, motor, cerebellar, and other clinical signs and symptoms. Neuroradiological findings typically involve the brainstem and cervical spinal cord. Adult-onset AxD has been described in diverse populations but is rare in Israel. We present a series of patients diagnosed with adult-onset AxD from three families, all of Jewish Syrian descent. Five patients (4 females) were diagnosed with adult-onset AxD due to the heterozygous mutation c.219G > A, p.Met73Ile in GFAP. Age at symptoms onset ranged from 48 to 61 years. Clinical characteristics were typical and involved progressive bulbar and gait disturbance, followed by pyramidal and cerebellar impairment, dysautonomia, and cognitive decline. Imaging findings included medullary and cervical spinal atrophy and mostly infratentorial white matter hyperintensities. A newly recognized cluster of adult-onset AxD in Jews of Syrian origin is presented. This disorder should be considered in differential diagnosis in appropriate circumstances. Genetic counselling for family members is required in order to discuss options for future family planning.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


