{"title":"Targeted sequencing analysis pipeline for species identification of human pathogenic fungi using long-read nanopore sequencing.","authors":"Nattapong Langsiri, Navaporn Worasilchai, Laszlo Irinyi, Piroon Jenjaroenpun, Thidathip Wongsurawat, Janet Jennifer Luangsa-Ard, Wieland Meyer, Ariya Chindamporn","doi":"10.1186/s43008-023-00125-6","DOIUrl":null,"url":null,"abstract":"<p><p>Among molecular-based techniques for fungal identification, Sanger sequencing of the primary universal fungal DNA barcode, the internal transcribed spacer (ITS) region (ITS1, 5.8S, ITS2), is commonly used in clinical routine laboratories due to its simplicity, universality, efficacy, and affordability for fungal species identification. However, Sanger sequencing fails to identify mixed ITS sequences in the case of mixed infections. To overcome this limitation, different high-throughput sequencing technologies have been explored. The nanopore-based technology is now one of the most promising long-read sequencing technologies on the market as it has the potential to sequence the full-length ITS region in a single read. In this study, we established a workflow for species identification using the sequences of the entire ITS region generated by nanopore sequencing of both pure yeast isolates and mocked mixed species reads generated with different scenarios. The species used in this study included Candida albicans (n = 2), Candida tropicalis (n = 1), Nakaseomyces glabratus (formerly Candida glabrata) (n = 1), Trichosporon asahii (n = 2), Pichia kudriavzevii (formerly Candida krusei) (n = 1), and Cryptococcus neoformans (n = 1). Comparing various methods to generate the consensus sequence for fungal species identification, the results from this study indicate that read clustering using a modified version of the NanoCLUST pipeline is more sensitive than Canu or VSEARCH, as it classified species accurately with a lower abundance cluster of reads (3% abundance compared to 10% with VSEARCH). The modified NanoCLUST also reduced the number of classified clusters compared to VSEARCH, making the subsequent BLAST+ analysis faster. Subsampling of the datasets, which reduces the size of the datasets by approximately tenfold, did not significantly affect the identification results in terms of the identified species name, percent identity, query coverage, percentage of reads in the classified cluster, and the number of clusters. The ability of the method to distinguish mixed species within sub-populations of large datasets has the potential to aid computer analysis by reducing the required processing power. The herein presented new sequence analysis pipeline will facilitate better interpretation of fungal sequence data for species identification.</p>","PeriodicalId":54345,"journal":{"name":"Ima Fungus","volume":"14 1","pages":"18"},"PeriodicalIF":6.2000,"publicationDate":"2023-09-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10483712/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Ima Fungus","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s43008-023-00125-6","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MYCOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
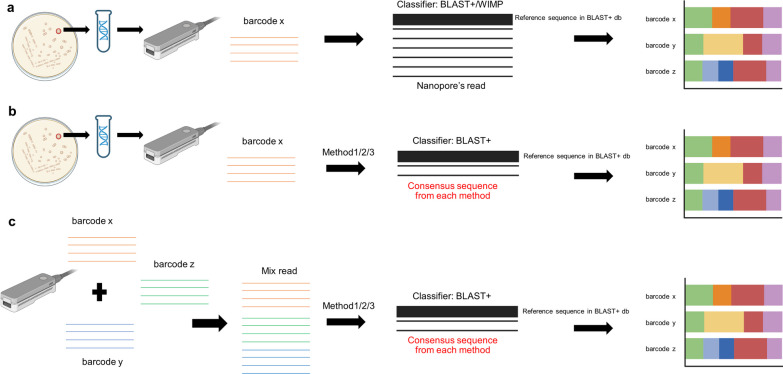
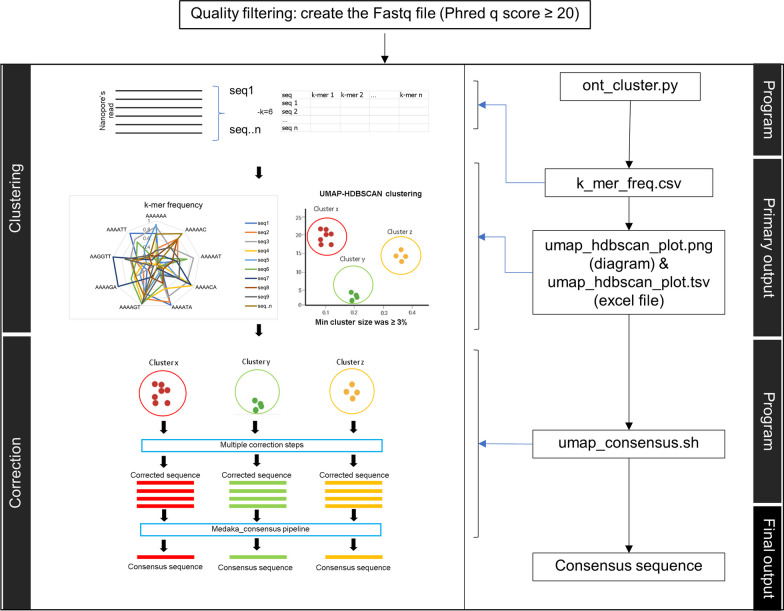
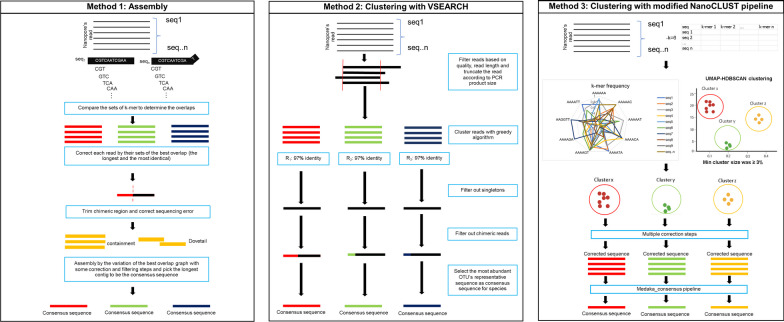
Among molecular-based techniques for fungal identification, Sanger sequencing of the primary universal fungal DNA barcode, the internal transcribed spacer (ITS) region (ITS1, 5.8S, ITS2), is commonly used in clinical routine laboratories due to its simplicity, universality, efficacy, and affordability for fungal species identification. However, Sanger sequencing fails to identify mixed ITS sequences in the case of mixed infections. To overcome this limitation, different high-throughput sequencing technologies have been explored. The nanopore-based technology is now one of the most promising long-read sequencing technologies on the market as it has the potential to sequence the full-length ITS region in a single read. In this study, we established a workflow for species identification using the sequences of the entire ITS region generated by nanopore sequencing of both pure yeast isolates and mocked mixed species reads generated with different scenarios. The species used in this study included Candida albicans (n = 2), Candida tropicalis (n = 1), Nakaseomyces glabratus (formerly Candida glabrata) (n = 1), Trichosporon asahii (n = 2), Pichia kudriavzevii (formerly Candida krusei) (n = 1), and Cryptococcus neoformans (n = 1). Comparing various methods to generate the consensus sequence for fungal species identification, the results from this study indicate that read clustering using a modified version of the NanoCLUST pipeline is more sensitive than Canu or VSEARCH, as it classified species accurately with a lower abundance cluster of reads (3% abundance compared to 10% with VSEARCH). The modified NanoCLUST also reduced the number of classified clusters compared to VSEARCH, making the subsequent BLAST+ analysis faster. Subsampling of the datasets, which reduces the size of the datasets by approximately tenfold, did not significantly affect the identification results in terms of the identified species name, percent identity, query coverage, percentage of reads in the classified cluster, and the number of clusters. The ability of the method to distinguish mixed species within sub-populations of large datasets has the potential to aid computer analysis by reducing the required processing power. The herein presented new sequence analysis pipeline will facilitate better interpretation of fungal sequence data for species identification.
Ima FungusAgricultural and Biological Sciences-Agricultural and Biological Sciences (miscellaneous)
CiteScore
11.00
自引率
3.70%
发文量
18
审稿时长
20 weeks
期刊介绍:
The flagship journal of the International Mycological Association. IMA Fungus is an international, peer-reviewed, open-access, full colour, fast-track journal. Papers on any aspect of mycology are considered, and published on-line with final pagination after proofs have been corrected; they are then effectively published under the International Code of Nomenclature for algae, fungi, and plants. The journal strongly supports good practice policies, and requires voucher specimens or cultures to be deposited in a public collection with an online database, DNA sequences in GenBank, alignments in TreeBASE, and validating information on new scientific names, including typifications, to be lodged in MycoBank. News, meeting reports, personalia, research news, correspondence, book news, and information on forthcoming international meetings are included in each issue

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


