Lianghao Si, Zhanjun Wang, Xu-Ying Li, Yang Song, Tingyan Yao, Erhe Xu, Xianling Wang, Chaodong Wang
{"title":"Novel mutations and molecular pathways identified in patients with brain iron accumulation disorders.","authors":"Lianghao Si, Zhanjun Wang, Xu-Ying Li, Yang Song, Tingyan Yao, Erhe Xu, Xianling Wang, Chaodong Wang","doi":"10.1007/s10048-023-00725-9","DOIUrl":null,"url":null,"abstract":"<p><p>Brain iron accumulation disorders (BIADs) are a group of diseases characterized by iron overload in deep gray matter nuclei, which is a common feature of neurodegenerative diseases. Although genetic factors have been reported to be one of the etiologies, much more details about the genetic background and molecular mechanism of BIADs remain unclear. This study aimed to illustrate the genetic characteristics of BIADs and clarify their molecular mechanisms. A total of 84 patients with BIADs were recruited from April 2018 to October 2022 at Xuanwu Hospital. Clinical characteristics including family history, consanguineous marriage history, and age at onset (AAO) were collected and assessed by two senior neurologists. Neuroimaging data were conducted for all the patients, including cranial magnetic resonance imaging (MRI) and susceptibility-weighted imaging (SWI). Whole-exome sequencing (WES) and capillary electrophoresis for detecting sequence mutation and trinucleotide repeat expansion, respectively, were conducted on all patients and part of their parents (whose samples were available). Variant pathogenicity was assessed according to the American College of Medical Genetics and Association for Molecular Pathology (ACMG/AMP). The NBIA and NBIA-like genes with mutations were included for bioinformatic analysis, using Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genome (KEGG). GO annotation and KEGG pathway analysis were performed on Metascape platform. In the 84 patients, 30 (35.7%) were found to carry mutations, among which 20 carried non-dynamic mutations (missense, stop-gained, frameshift, inframe, and exonic deletion) and 10 carried repeat expansion mutations. Compared with sporadic cases, familial cases had more genetic variants (non-dynamic mutation: P=0.025, dynamic mutation: P=0.003). AAO was 27.85±10.42 years in cases with non-dynamic mutations, which was significantly younger than those without mutations (43.13±17.17, t=3.724, P<0.001) and those with repeated expansions (45.40±8.90, t=4.550, P<0.001). Bioinformatic analysis suggested that genes in lipid metabolism, autophagy, mitochondria regulation, and ferroptosis pathways are more likely to be involved in the pathogenesis of BIADs. This study broadens the genetic spectrum of BIADs and has important implications in genetic counselling and clinical diagnosis. Patients diagnosed as BIADs with early AAO and family history are more likely to carry mutations. Bioinformatic analysis provides new insights into the molecular pathogenesis of BIADs, which may shed lights on the therapeutic strategy for neurodegenerative diseases.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":" ","pages":"231-241"},"PeriodicalIF":1.6000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00725-9","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/7/15 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
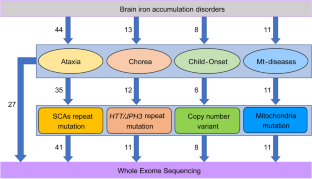
Brain iron accumulation disorders (BIADs) are a group of diseases characterized by iron overload in deep gray matter nuclei, which is a common feature of neurodegenerative diseases. Although genetic factors have been reported to be one of the etiologies, much more details about the genetic background and molecular mechanism of BIADs remain unclear. This study aimed to illustrate the genetic characteristics of BIADs and clarify their molecular mechanisms. A total of 84 patients with BIADs were recruited from April 2018 to October 2022 at Xuanwu Hospital. Clinical characteristics including family history, consanguineous marriage history, and age at onset (AAO) were collected and assessed by two senior neurologists. Neuroimaging data were conducted for all the patients, including cranial magnetic resonance imaging (MRI) and susceptibility-weighted imaging (SWI). Whole-exome sequencing (WES) and capillary electrophoresis for detecting sequence mutation and trinucleotide repeat expansion, respectively, were conducted on all patients and part of their parents (whose samples were available). Variant pathogenicity was assessed according to the American College of Medical Genetics and Association for Molecular Pathology (ACMG/AMP). The NBIA and NBIA-like genes with mutations were included for bioinformatic analysis, using Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genome (KEGG). GO annotation and KEGG pathway analysis were performed on Metascape platform. In the 84 patients, 30 (35.7%) were found to carry mutations, among which 20 carried non-dynamic mutations (missense, stop-gained, frameshift, inframe, and exonic deletion) and 10 carried repeat expansion mutations. Compared with sporadic cases, familial cases had more genetic variants (non-dynamic mutation: P=0.025, dynamic mutation: P=0.003). AAO was 27.85±10.42 years in cases with non-dynamic mutations, which was significantly younger than those without mutations (43.13±17.17, t=3.724, P<0.001) and those with repeated expansions (45.40±8.90, t=4.550, P<0.001). Bioinformatic analysis suggested that genes in lipid metabolism, autophagy, mitochondria regulation, and ferroptosis pathways are more likely to be involved in the pathogenesis of BIADs. This study broadens the genetic spectrum of BIADs and has important implications in genetic counselling and clinical diagnosis. Patients diagnosed as BIADs with early AAO and family history are more likely to carry mutations. Bioinformatic analysis provides new insights into the molecular pathogenesis of BIADs, which may shed lights on the therapeutic strategy for neurodegenerative diseases.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


