Mary P Choules, Peiying Zuo, Yukio Otsuka, Amit Garg, Mei Tang, Peter Bonate
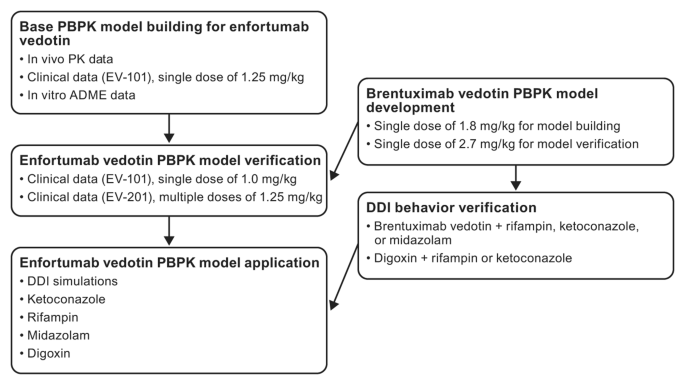
{"title":"Physiologically based pharmacokinetic model to predict drug-drug interactions with the antibody-drug conjugate enfortumab vedotin.","authors":"Mary P Choules, Peiying Zuo, Yukio Otsuka, Amit Garg, Mei Tang, Peter Bonate","doi":"10.1007/s10928-023-09877-5","DOIUrl":null,"url":null,"abstract":"<p><p>Enfortumab vedotin is an antibody-drug conjugate (ADC) comprised of a Nectin-4-directed antibody and monomethyl auristatin E (MMAE), which is primarily eliminated through P-glycoprotein (P-gp)-mediated excretion and cytochrome P450 3A4 (CYP3A4)-mediated metabolism. A physiologically based pharmacokinetic (PBPK) model was developed to predict effects of combined P-gp with CYP3A4 inhibitor/inducer (ketoconazole/rifampin) on MMAE exposure when coadministered with enfortumab vedotin and study enfortumab vedotin with CYP3A4 (midazolam) and P-gp (digoxin) substrate exposure. A PBPK model was built for enfortumab vedotin and unconjugated MMAE using the PBPK simulator ADC module. A similar model was developed with brentuximab vedotin, an ADC with the same valine-citrulline-MMAE linker as enfortumab vedotin, for MMAE drug-drug interaction (DDI) verification using clinical data. The DDI simulation predicted a less-than-2-fold increase in MMAE exposure with enfortumab vedotin plus ketoconazole (MMAE geometric mean ratio [GMR] for maximum concentration [C<sub>max</sub>], 1.15; GMR for area under the time-concentration curve from time 0 to last quantifiable concentration [AUC<sub>last</sub>], 1.38). Decreased MMAE exposure above 50% but below 80% was observed with enfortumab vedotin plus rifampin (MMAE GMR C<sub>max</sub>, 0.72; GMR AUC<sub>last</sub>, 0.47). No effect of enfortumab vedotin on midazolam or digoxin systemic exposure was predicted. Results suggest that combination enfortumab vedotin, P-gp, and a CYP3A4 inhibitor may result in increased MMAE exposure and patients should be monitored for potential adverse effects. Combination P-gp and a CYP3A4 inducer may result in decreased MMAE exposure. No exposure change is expected for CYP3A4 or P-gp substrates when combined with enfortumab vedotin.ClinicalTrials.gov identifier Not applicable.</p>","PeriodicalId":16851,"journal":{"name":"Journal of Pharmacokinetics and Pharmacodynamics","volume":" ","pages":"417-428"},"PeriodicalIF":2.8000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11576838/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Pharmacokinetics and Pharmacodynamics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10928-023-09877-5","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/26 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
Enfortumab vedotin is an antibody-drug conjugate (ADC) comprised of a Nectin-4-directed antibody and monomethyl auristatin E (MMAE), which is primarily eliminated through P-glycoprotein (P-gp)-mediated excretion and cytochrome P450 3A4 (CYP3A4)-mediated metabolism. A physiologically based pharmacokinetic (PBPK) model was developed to predict effects of combined P-gp with CYP3A4 inhibitor/inducer (ketoconazole/rifampin) on MMAE exposure when coadministered with enfortumab vedotin and study enfortumab vedotin with CYP3A4 (midazolam) and P-gp (digoxin) substrate exposure. A PBPK model was built for enfortumab vedotin and unconjugated MMAE using the PBPK simulator ADC module. A similar model was developed with brentuximab vedotin, an ADC with the same valine-citrulline-MMAE linker as enfortumab vedotin, for MMAE drug-drug interaction (DDI) verification using clinical data. The DDI simulation predicted a less-than-2-fold increase in MMAE exposure with enfortumab vedotin plus ketoconazole (MMAE geometric mean ratio [GMR] for maximum concentration [Cmax], 1.15; GMR for area under the time-concentration curve from time 0 to last quantifiable concentration [AUClast], 1.38). Decreased MMAE exposure above 50% but below 80% was observed with enfortumab vedotin plus rifampin (MMAE GMR Cmax, 0.72; GMR AUClast, 0.47). No effect of enfortumab vedotin on midazolam or digoxin systemic exposure was predicted. Results suggest that combination enfortumab vedotin, P-gp, and a CYP3A4 inhibitor may result in increased MMAE exposure and patients should be monitored for potential adverse effects. Combination P-gp and a CYP3A4 inducer may result in decreased MMAE exposure. No exposure change is expected for CYP3A4 or P-gp substrates when combined with enfortumab vedotin.ClinicalTrials.gov identifier Not applicable.
期刊介绍:
Broadly speaking, the Journal of Pharmacokinetics and Pharmacodynamics covers the area of pharmacometrics. The journal is devoted to illustrating the importance of pharmacokinetics, pharmacodynamics, and pharmacometrics in drug development, clinical care, and the understanding of drug action. The journal publishes on a variety of topics related to pharmacometrics, including, but not limited to, clinical, experimental, and theoretical papers examining the kinetics of drug disposition and effects of drug action in humans, animals, in vitro, or in silico; modeling and simulation methodology, including optimal design; precision medicine; systems pharmacology; and mathematical pharmacology (including computational biology, bioengineering, and biophysics related to pharmacology, pharmacokinetics, orpharmacodynamics). Clinical papers that include population pharmacokinetic-pharmacodynamic relationships are welcome. The journal actively invites and promotes up-and-coming areas of pharmacometric research, such as real-world evidence, quality of life analyses, and artificial intelligence. The Journal of Pharmacokinetics and Pharmacodynamics is an official journal of the International Society of Pharmacometrics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


