Christiana O Shobo, Daniel G Amoako, Mushal Allam, Arshad Ismail, Sabiha Y Essack, Linda A Bester
{"title":"A Genomic Snapshot of Antibiotic-Resistant<i>Enterococcus faecalis</i> within Public Hospital Environments in South Africa.","authors":"Christiana O Shobo, Daniel G Amoako, Mushal Allam, Arshad Ismail, Sabiha Y Essack, Linda A Bester","doi":"10.1155/2023/6639983","DOIUrl":null,"url":null,"abstract":"<p><p>Enterococci are among the most common opportunistic hospital pathogens. This study used whole-genome sequencing (WGS) and bioinformatics to determine the antibiotic resistome, mobile genetic elements, clone and phylogenetic relationship of <i>Enterococcus faecalis</i> isolated from hospital environments in South Africa. This study was carried out from September to November 2017. Isolates were recovered from 11 frequently touched sites by patients and healthcare workers in different wards at 4 levels of healthcare (A, B, C, and D) in Durban, South Africa. Out of the 245 identified <i>E. faecalis</i> isolates, 38 isolates underwent whole-genome sequencing (WGS) on the Illumina MiSeq platform, following microbial identification and antibiotic susceptibility tests. The <i>tet(M)</i> (31/38, 82%) and <i>erm(C)</i> (16/38, 42%) genes were the most common antibiotic-resistant genes found in isolates originating from different hospital environments which corroborated with their antibiotic resistance phenotypes. The isolates harboured mobile genetic elements consisting of plasmids (<i>n</i> = 11) and prophages (<i>n</i> = 14) that were mostly clone-specific. Of note, a large number of insertion sequence (IS) families were found on the IS3 (55%), IS5 (42%), IS1595 (40%), and Tn3 transposons the most predominant. Microbial typing using WGS data revealed 15 clones with 6 major sequence types (ST) belonging to ST16 (<i>n</i> = 7), ST40 (<i>n</i> = 6), ST21 (<i>n</i> = 5), ST126 (<i>n</i> = 3), ST23 (<i>n</i> = 3), and ST386 (<i>n</i> = 3). Phylogenomic analysis showed that the major clones were mostly conserved within specific hospital environments. However, further metadata insights revealed the complex intraclonal spread of these <i>E. faecalis</i> major clones between the sampling sites within each specific hospital setting. The results of these genomic analyses will offer insights into antibiotic-resistant<i>E. faecalis</i> in hospital environments relevant to the design of optimal infection prevention strategies in hospital settings.</p>","PeriodicalId":44052,"journal":{"name":"Global Health Epidemiology and Genomics","volume":"2023 ","pages":"6639983"},"PeriodicalIF":1.5000,"publicationDate":"2023-06-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10279497/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Global Health Epidemiology and Genomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2023/6639983","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"PUBLIC, ENVIRONMENTAL & OCCUPATIONAL HEALTH","Score":null,"Total":0}
引用次数: 0
Abstract
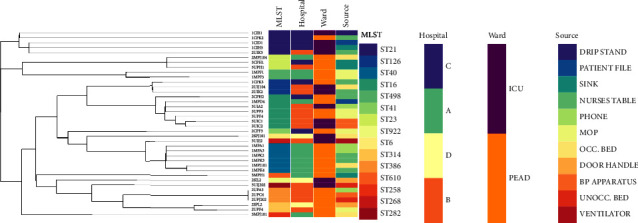
Enterococci are among the most common opportunistic hospital pathogens. This study used whole-genome sequencing (WGS) and bioinformatics to determine the antibiotic resistome, mobile genetic elements, clone and phylogenetic relationship of Enterococcus faecalis isolated from hospital environments in South Africa. This study was carried out from September to November 2017. Isolates were recovered from 11 frequently touched sites by patients and healthcare workers in different wards at 4 levels of healthcare (A, B, C, and D) in Durban, South Africa. Out of the 245 identified E. faecalis isolates, 38 isolates underwent whole-genome sequencing (WGS) on the Illumina MiSeq platform, following microbial identification and antibiotic susceptibility tests. The tet(M) (31/38, 82%) and erm(C) (16/38, 42%) genes were the most common antibiotic-resistant genes found in isolates originating from different hospital environments which corroborated with their antibiotic resistance phenotypes. The isolates harboured mobile genetic elements consisting of plasmids (n = 11) and prophages (n = 14) that were mostly clone-specific. Of note, a large number of insertion sequence (IS) families were found on the IS3 (55%), IS5 (42%), IS1595 (40%), and Tn3 transposons the most predominant. Microbial typing using WGS data revealed 15 clones with 6 major sequence types (ST) belonging to ST16 (n = 7), ST40 (n = 6), ST21 (n = 5), ST126 (n = 3), ST23 (n = 3), and ST386 (n = 3). Phylogenomic analysis showed that the major clones were mostly conserved within specific hospital environments. However, further metadata insights revealed the complex intraclonal spread of these E. faecalis major clones between the sampling sites within each specific hospital setting. The results of these genomic analyses will offer insights into antibiotic-resistantE. faecalis in hospital environments relevant to the design of optimal infection prevention strategies in hospital settings.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


