Jeremiah M Afolabi, Praghalathan Kanthakumar, Jada D Williams, Ravi Kumar, Hitesh Soni, Adebowale Adebiyi
{"title":"Post-injury Inhibition of Endothelin-1 Dependent Renal Vasoregulation Mitigates Rhabdomyolysis-Induced Acute Kidney Injury.","authors":"Jeremiah M Afolabi, Praghalathan Kanthakumar, Jada D Williams, Ravi Kumar, Hitesh Soni, Adebowale Adebiyi","doi":"10.1093/function/zqad022","DOIUrl":null,"url":null,"abstract":"<p><p>In patients with rhabdomyolysis, the overwhelming release of myoglobin into the circulation is the primary cause of kidney injury. Myoglobin causes direct kidney injury as well as severe renal vasoconstriction. An increase in renal vascular resistance (RVR) results in renal blood flow (RBF) and glomerular filtration rate (GFR) reduction, tubular injury, and acute kidney injury (AKI). The mechanisms that underlie rhabdomyolysis-induced AKI are not fully understood but may involve the local production of vasoactive mediators in the kidney. Studies have shown that myoglobin stimulates endothelin-1 (ET-1) production in glomerular mesangial cells. Circulating ET-1 is also increased in rats subjected to glycerol-induced rhabdomyolysis. However, the upstream mechanisms of ET-1 production and downstream effectors of ET-1 actions in rhabdomyolysis-induced AKI remain unclear. Vasoactive ET-1 is generated by ET converting enzyme 1 (ECE-1)-induced proteolytic processing of inactive big ET to biologically active peptides. The downstream ion channel effectors of ET-1-induced vasoregulation include the transient receptor potential cation channel, subfamily C member 3 (TRPC3). This study demonstrates that glycerol-induced rhabdomyolysis in Wistar rats promotes ECE-1-dependent ET-1 production, RVR increase, GFR decrease, and AKI. Rhabdomyolysis-induced increases in RVR and AKI in the rats were attenuated by post-injury pharmacological inhibition of ECE-1, ET receptors, and TRPC3 channels. CRISPR/Cas9-mediated knockout of TRPC3 channels attenuated ET-1-induced renal vascular reactivity and rhabdomyolysis-induced AKI. These findings suggest that ECE-1-driven ET-1 production and downstream activation of TRPC3-dependent renal vasoconstriction contribute to rhabdomyolysis-induced AKI. Hence, post-injury inhibition of ET-1-mediated renal vasoregulation may provide therapeutic targets for rhabdomyolysis-induced AKI.</p>","PeriodicalId":73119,"journal":{"name":"Function (Oxford, England)","volume":"4 4","pages":"zqad022"},"PeriodicalIF":3.8000,"publicationDate":"2023-05-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/06/c0/zqad022.PMC10278989.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Function (Oxford, England)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/function/zqad022","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
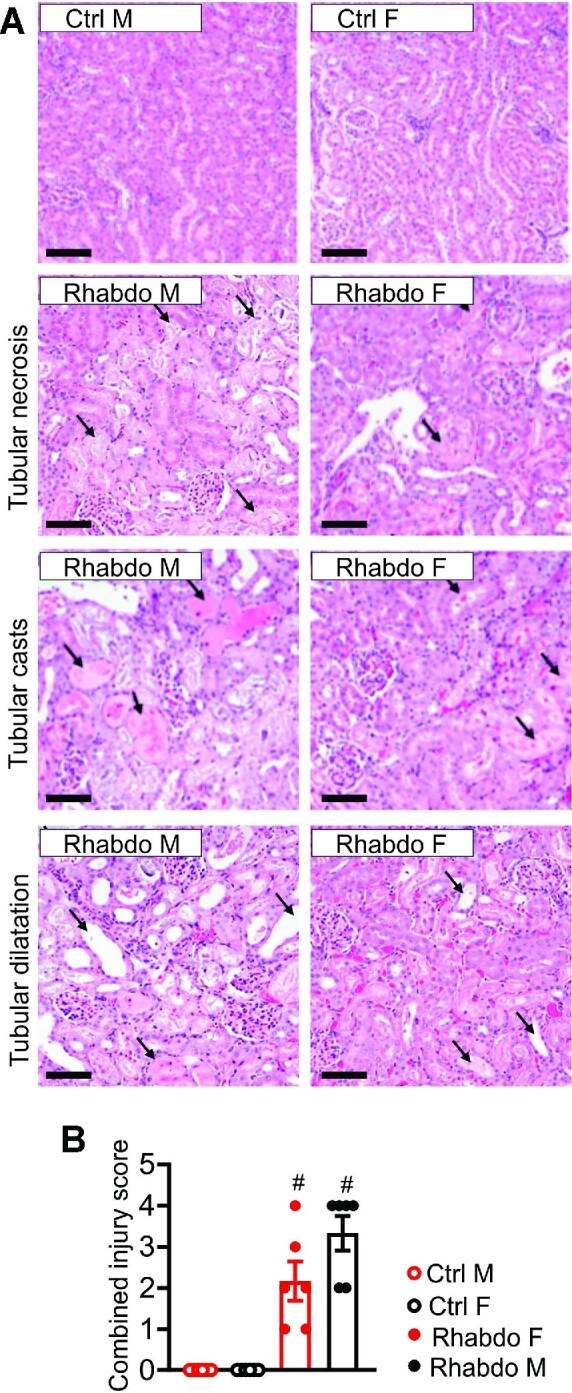
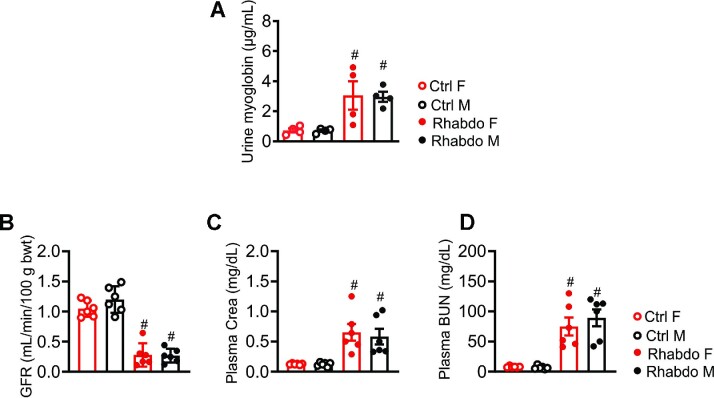
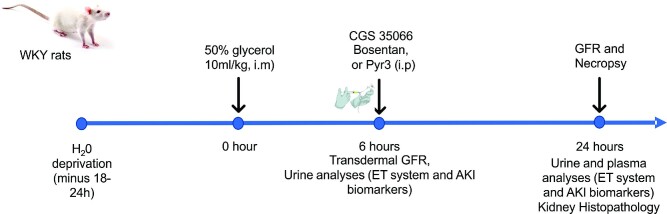
In patients with rhabdomyolysis, the overwhelming release of myoglobin into the circulation is the primary cause of kidney injury. Myoglobin causes direct kidney injury as well as severe renal vasoconstriction. An increase in renal vascular resistance (RVR) results in renal blood flow (RBF) and glomerular filtration rate (GFR) reduction, tubular injury, and acute kidney injury (AKI). The mechanisms that underlie rhabdomyolysis-induced AKI are not fully understood but may involve the local production of vasoactive mediators in the kidney. Studies have shown that myoglobin stimulates endothelin-1 (ET-1) production in glomerular mesangial cells. Circulating ET-1 is also increased in rats subjected to glycerol-induced rhabdomyolysis. However, the upstream mechanisms of ET-1 production and downstream effectors of ET-1 actions in rhabdomyolysis-induced AKI remain unclear. Vasoactive ET-1 is generated by ET converting enzyme 1 (ECE-1)-induced proteolytic processing of inactive big ET to biologically active peptides. The downstream ion channel effectors of ET-1-induced vasoregulation include the transient receptor potential cation channel, subfamily C member 3 (TRPC3). This study demonstrates that glycerol-induced rhabdomyolysis in Wistar rats promotes ECE-1-dependent ET-1 production, RVR increase, GFR decrease, and AKI. Rhabdomyolysis-induced increases in RVR and AKI in the rats were attenuated by post-injury pharmacological inhibition of ECE-1, ET receptors, and TRPC3 channels. CRISPR/Cas9-mediated knockout of TRPC3 channels attenuated ET-1-induced renal vascular reactivity and rhabdomyolysis-induced AKI. These findings suggest that ECE-1-driven ET-1 production and downstream activation of TRPC3-dependent renal vasoconstriction contribute to rhabdomyolysis-induced AKI. Hence, post-injury inhibition of ET-1-mediated renal vasoregulation may provide therapeutic targets for rhabdomyolysis-induced AKI.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


