David Teschner, David Gomez-Zepeda, Arthur Declercq, Mateusz K Łącki, Seymen Avci, Konstantin Bob, Ute Distler, Thomas Michna, Lennart Martens, Stefan Tenzer, Andreas Hildebrandt
{"title":"Ionmob:用于预测肽碰撞横截面值的Python包。","authors":"David Teschner, David Gomez-Zepeda, Arthur Declercq, Mateusz K Łącki, Seymen Avci, Konstantin Bob, Ute Distler, Thomas Michna, Lennart Martens, Stefan Tenzer, Andreas Hildebrandt","doi":"10.1093/bioinformatics/btad486","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Including ion mobility separation (IMS) into mass spectrometry proteomics experiments is useful to improve coverage and throughput. Many IMS devices enable linking experimentally derived mobility of an ion to its collisional cross-section (CCS), a highly reproducible physicochemical property dependent on the ion's mass, charge and conformation in the gas phase. Thus, known peptide ion mobilities can be used to tailor acquisition methods or to refine database search results. The large space of potential peptide sequences, driven also by posttranslational modifications of amino acids, motivates an in silico predictor for peptide CCS. Recent studies explored the general performance of varying machine-learning techniques, however, the workflow engineering part was of secondary importance. For the sake of applicability, such a tool should be generic, data driven, and offer the possibility to be easily adapted to individual workflows for experimental design and data processing.</p><p><strong>Results: </strong>We created ionmob, a Python-based framework for data preparation, training, and prediction of collisional cross-section values of peptides. It is easily customizable and includes a set of pretrained, ready-to-use models and preprocessing routines for training and inference. Using a set of ≈21 000 unique phosphorylated peptides and ≈17 000 MHC ligand sequences and charge state pairs, we expand upon the space of peptides that can be integrated into CCS prediction. Lastly, we investigate the applicability of in silico predicted CCS to increase confidence in identified peptides by applying methods of re-scoring and demonstrate that predicted CCS values complement existing predictors for that task.</p><p><strong>Availability and implementation: </strong>The Python package is available at github: https://github.com/theGreatHerrLebert/ionmob.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":" ","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10521631/pdf/","citationCount":"0","resultStr":"{\"title\":\"Ionmob: a Python package for prediction of peptide collisional cross-section values.\",\"authors\":\"David Teschner, David Gomez-Zepeda, Arthur Declercq, Mateusz K Łącki, Seymen Avci, Konstantin Bob, Ute Distler, Thomas Michna, Lennart Martens, Stefan Tenzer, Andreas Hildebrandt\",\"doi\":\"10.1093/bioinformatics/btad486\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>Including ion mobility separation (IMS) into mass spectrometry proteomics experiments is useful to improve coverage and throughput. Many IMS devices enable linking experimentally derived mobility of an ion to its collisional cross-section (CCS), a highly reproducible physicochemical property dependent on the ion's mass, charge and conformation in the gas phase. Thus, known peptide ion mobilities can be used to tailor acquisition methods or to refine database search results. The large space of potential peptide sequences, driven also by posttranslational modifications of amino acids, motivates an in silico predictor for peptide CCS. Recent studies explored the general performance of varying machine-learning techniques, however, the workflow engineering part was of secondary importance. For the sake of applicability, such a tool should be generic, data driven, and offer the possibility to be easily adapted to individual workflows for experimental design and data processing.</p><p><strong>Results: </strong>We created ionmob, a Python-based framework for data preparation, training, and prediction of collisional cross-section values of peptides. It is easily customizable and includes a set of pretrained, ready-to-use models and preprocessing routines for training and inference. Using a set of ≈21 000 unique phosphorylated peptides and ≈17 000 MHC ligand sequences and charge state pairs, we expand upon the space of peptides that can be integrated into CCS prediction. Lastly, we investigate the applicability of in silico predicted CCS to increase confidence in identified peptides by applying methods of re-scoring and demonstrate that predicted CCS values complement existing predictors for that task.</p><p><strong>Availability and implementation: </strong>The Python package is available at github: https://github.com/theGreatHerrLebert/ionmob.</p>\",\"PeriodicalId\":8903,\"journal\":{\"name\":\"Bioinformatics\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2023-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10521631/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bioinformatics/btad486\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad486","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Ionmob: a Python package for prediction of peptide collisional cross-section values.
Motivation: Including ion mobility separation (IMS) into mass spectrometry proteomics experiments is useful to improve coverage and throughput. Many IMS devices enable linking experimentally derived mobility of an ion to its collisional cross-section (CCS), a highly reproducible physicochemical property dependent on the ion's mass, charge and conformation in the gas phase. Thus, known peptide ion mobilities can be used to tailor acquisition methods or to refine database search results. The large space of potential peptide sequences, driven also by posttranslational modifications of amino acids, motivates an in silico predictor for peptide CCS. Recent studies explored the general performance of varying machine-learning techniques, however, the workflow engineering part was of secondary importance. For the sake of applicability, such a tool should be generic, data driven, and offer the possibility to be easily adapted to individual workflows for experimental design and data processing.
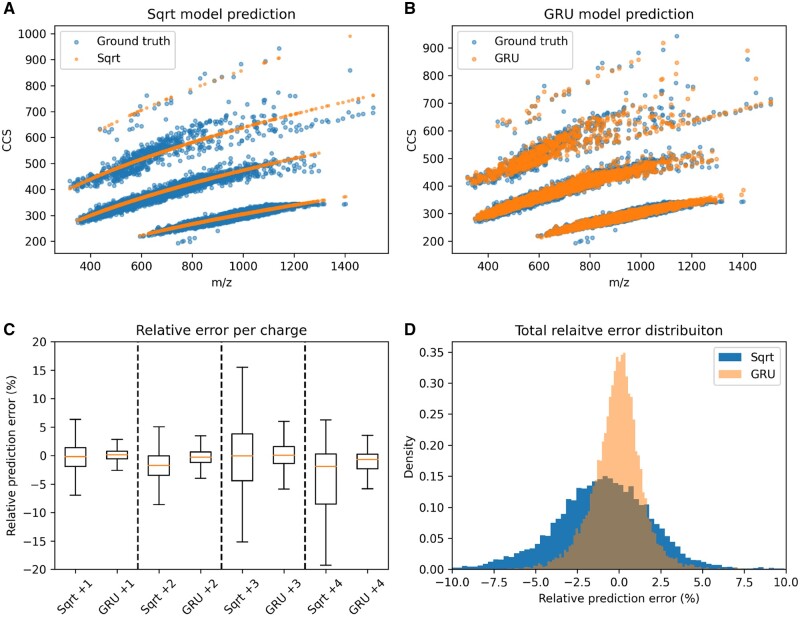
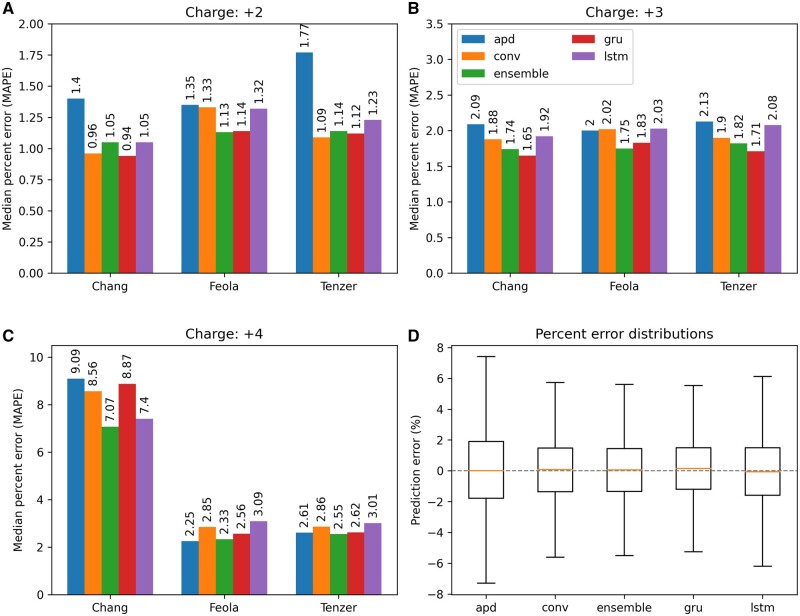
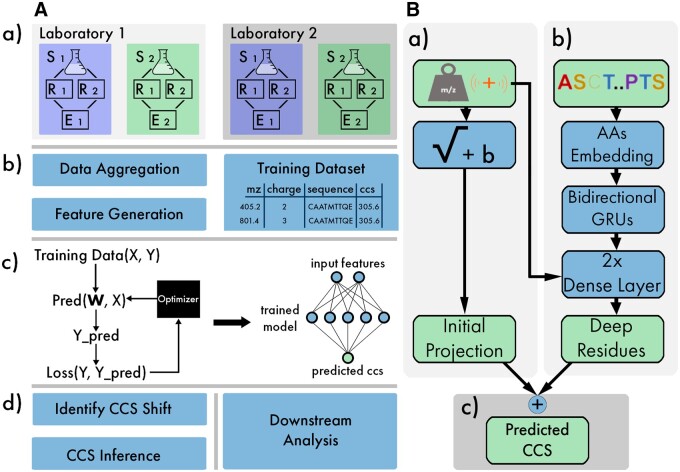
Results: We created ionmob, a Python-based framework for data preparation, training, and prediction of collisional cross-section values of peptides. It is easily customizable and includes a set of pretrained, ready-to-use models and preprocessing routines for training and inference. Using a set of ≈21 000 unique phosphorylated peptides and ≈17 000 MHC ligand sequences and charge state pairs, we expand upon the space of peptides that can be integrated into CCS prediction. Lastly, we investigate the applicability of in silico predicted CCS to increase confidence in identified peptides by applying methods of re-scoring and demonstrate that predicted CCS values complement existing predictors for that task.
Availability and implementation: The Python package is available at github: https://github.com/theGreatHerrLebert/ionmob.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


