{"title":"scRNA-seq中的基因表达与转录物3'端的常见基序相关。","authors":"Xinling Li, Greg Gibson, Peng Qiu","doi":"10.3389/fbinf.2023.1120290","DOIUrl":null,"url":null,"abstract":"<p><p>One important characteristic of single-cell RNA sequencing (scRNA-seq) data is its high sparsity, where the gene-cell count data matrix contains high proportion of zeros. The sparsity has motivated widespread discussions on dropouts and missing data, as well as imputation algorithms of scRNA-seq analysis. Here, we aim to investigate whether there exist genes that are more prone to be under-detected in scRNA-seq, and if yes, what commonalities those genes may share. From public data sources, we gathered paired bulk RNA-seq and scRNA-seq data from 53 human samples, which were generated in diverse biological contexts. We derived pseudo-bulk gene expression by averaging the scRNA-seq data across cells. Comparisons of the paired bulk and pseudo-bulk gene expression profiles revealed that there indeed exists a collection of genes that are frequently under-detected in scRNA-seq compared to bulk RNA-seq. This result was robust to randomization when unpaired bulk and pseudo-bulk gene expression profiles were compared. We performed motif search to the last 350 bp of the identified genes, and observed an enrichment of poly(T) motif. The poly(T) motif toward the tails of those genes may be able to form hairpin structures with the poly(A) tails of their mRNA transcripts, making it difficult for their mRNA transcripts to be captured during scRNA-seq library preparation, which is a mechanistic conjecture of why certain genes may be more prone to be under-detected in scRNA-seq.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"3 ","pages":"1120290"},"PeriodicalIF":2.8000,"publicationDate":"2023-05-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10226423/pdf/","citationCount":"0","resultStr":"{\"title\":\"Gene representation in scRNA-seq is correlated with common motifs at the 3' end of transcripts.\",\"authors\":\"Xinling Li, Greg Gibson, Peng Qiu\",\"doi\":\"10.3389/fbinf.2023.1120290\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>One important characteristic of single-cell RNA sequencing (scRNA-seq) data is its high sparsity, where the gene-cell count data matrix contains high proportion of zeros. The sparsity has motivated widespread discussions on dropouts and missing data, as well as imputation algorithms of scRNA-seq analysis. Here, we aim to investigate whether there exist genes that are more prone to be under-detected in scRNA-seq, and if yes, what commonalities those genes may share. From public data sources, we gathered paired bulk RNA-seq and scRNA-seq data from 53 human samples, which were generated in diverse biological contexts. We derived pseudo-bulk gene expression by averaging the scRNA-seq data across cells. Comparisons of the paired bulk and pseudo-bulk gene expression profiles revealed that there indeed exists a collection of genes that are frequently under-detected in scRNA-seq compared to bulk RNA-seq. This result was robust to randomization when unpaired bulk and pseudo-bulk gene expression profiles were compared. We performed motif search to the last 350 bp of the identified genes, and observed an enrichment of poly(T) motif. The poly(T) motif toward the tails of those genes may be able to form hairpin structures with the poly(A) tails of their mRNA transcripts, making it difficult for their mRNA transcripts to be captured during scRNA-seq library preparation, which is a mechanistic conjecture of why certain genes may be more prone to be under-detected in scRNA-seq.</p>\",\"PeriodicalId\":73066,\"journal\":{\"name\":\"Frontiers in bioinformatics\",\"volume\":\"3 \",\"pages\":\"1120290\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2023-05-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10226423/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Frontiers in bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3389/fbinf.2023.1120290\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2023.1120290","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Gene representation in scRNA-seq is correlated with common motifs at the 3' end of transcripts.
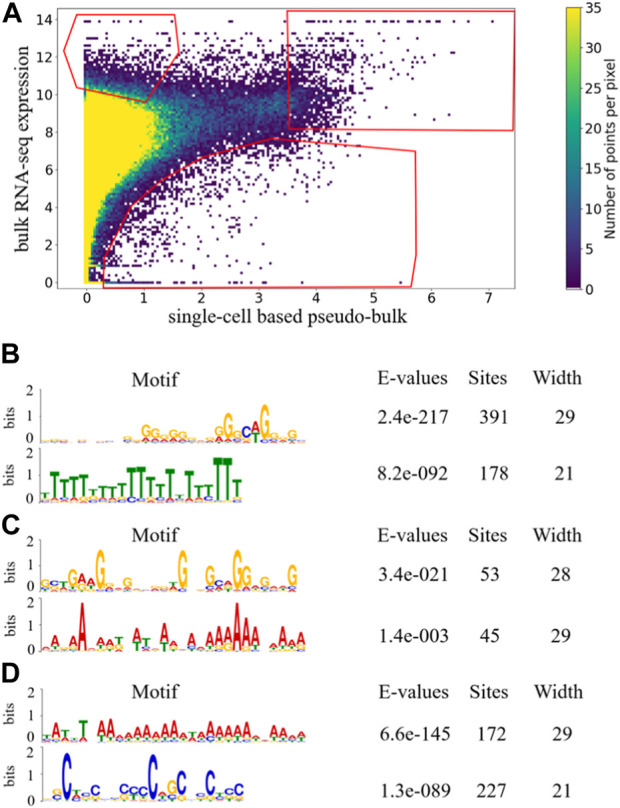
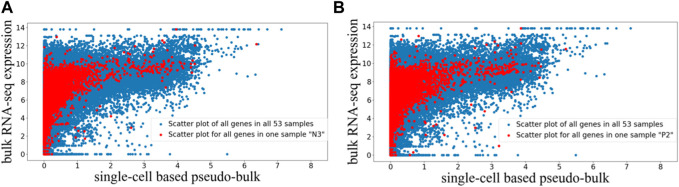
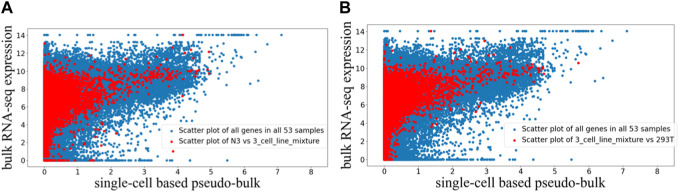
One important characteristic of single-cell RNA sequencing (scRNA-seq) data is its high sparsity, where the gene-cell count data matrix contains high proportion of zeros. The sparsity has motivated widespread discussions on dropouts and missing data, as well as imputation algorithms of scRNA-seq analysis. Here, we aim to investigate whether there exist genes that are more prone to be under-detected in scRNA-seq, and if yes, what commonalities those genes may share. From public data sources, we gathered paired bulk RNA-seq and scRNA-seq data from 53 human samples, which were generated in diverse biological contexts. We derived pseudo-bulk gene expression by averaging the scRNA-seq data across cells. Comparisons of the paired bulk and pseudo-bulk gene expression profiles revealed that there indeed exists a collection of genes that are frequently under-detected in scRNA-seq compared to bulk RNA-seq. This result was robust to randomization when unpaired bulk and pseudo-bulk gene expression profiles were compared. We performed motif search to the last 350 bp of the identified genes, and observed an enrichment of poly(T) motif. The poly(T) motif toward the tails of those genes may be able to form hairpin structures with the poly(A) tails of their mRNA transcripts, making it difficult for their mRNA transcripts to be captured during scRNA-seq library preparation, which is a mechanistic conjecture of why certain genes may be more prone to be under-detected in scRNA-seq.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


