Jingxuan Chen, Preston J Basting, Shunhua Han, David J Garfinkel, Casey M Bergman
{"title":"用McClintock 2对转座元件检测器的可重复性评价指导了酵母中Ty插入模式的准确推断。","authors":"Jingxuan Chen, Preston J Basting, Shunhua Han, David J Garfinkel, Casey M Bergman","doi":"10.1186/s13100-023-00296-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Many computational methods have been developed to detect non-reference transposable element (TE) insertions using short-read whole genome sequencing data. The diversity and complexity of such methods often present challenges to new users seeking to reproducibly install, execute, or evaluate multiple TE insertion detectors.</p><p><strong>Results: </strong>We previously developed the McClintock meta-pipeline to facilitate the installation, execution, and evaluation of six first-generation short-read TE detectors. Here, we report a completely re-implemented version of McClintock written in Python using Snakemake and Conda that improves its installation, error handling, speed, stability, and extensibility. McClintock 2 now includes 12 short-read TE detectors, auxiliary pre-processing and analysis modules, interactive HTML reports, and a simulation framework to reproducibly evaluate the accuracy of component TE detectors. When applied to the model microbial eukaryote Saccharomyces cerevisiae, we find substantial variation in the ability of McClintock 2 components to identify the precise locations of non-reference TE insertions, with RelocaTE2 showing the highest recall and precision in simulated data. We find that RelocaTE2, TEMP, TEMP2 and TEBreak provide consistent estimates of [Formula: see text]50 non-reference TE insertions per strain and that Ty2 has the highest number of non-reference TE insertions in a species-wide panel of [Formula: see text]1000 yeast genomes. Finally, we show that best-in-class predictors for yeast applied to resequencing data have sufficient resolution to reveal a dyad pattern of integration in nucleosome-bound regions upstream of yeast tRNA genes for Ty1, Ty2, and Ty4, allowing us to extend knowledge about fine-scale target preferences revealed previously for experimentally-induced Ty1 insertions to spontaneous insertions for other copia-superfamily retrotransposons in yeast.</p><p><strong>Conclusion: </strong>McClintock ( https://github.com/bergmanlab/mcclintock/ ) provides a user-friendly pipeline for the identification of TEs in short-read WGS data using multiple TE detectors, which should benefit researchers studying TE insertion variation in a wide range of different organisms. Application of the improved McClintock system to simulated and empirical yeast genome data reveals best-in-class methods and novel biological insights for one of the most widely-studied model eukaryotes and provides a paradigm for evaluating and selecting non-reference TE detectors in other species.</p>","PeriodicalId":18854,"journal":{"name":"Mobile DNA","volume":"14 1","pages":"8"},"PeriodicalIF":4.7000,"publicationDate":"2023-07-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10347736/pdf/","citationCount":"1","resultStr":"{\"title\":\"Reproducible evaluation of transposable element detectors with McClintock 2 guides accurate inference of Ty insertion patterns in yeast.\",\"authors\":\"Jingxuan Chen, Preston J Basting, Shunhua Han, David J Garfinkel, Casey M Bergman\",\"doi\":\"10.1186/s13100-023-00296-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Many computational methods have been developed to detect non-reference transposable element (TE) insertions using short-read whole genome sequencing data. The diversity and complexity of such methods often present challenges to new users seeking to reproducibly install, execute, or evaluate multiple TE insertion detectors.</p><p><strong>Results: </strong>We previously developed the McClintock meta-pipeline to facilitate the installation, execution, and evaluation of six first-generation short-read TE detectors. Here, we report a completely re-implemented version of McClintock written in Python using Snakemake and Conda that improves its installation, error handling, speed, stability, and extensibility. McClintock 2 now includes 12 short-read TE detectors, auxiliary pre-processing and analysis modules, interactive HTML reports, and a simulation framework to reproducibly evaluate the accuracy of component TE detectors. When applied to the model microbial eukaryote Saccharomyces cerevisiae, we find substantial variation in the ability of McClintock 2 components to identify the precise locations of non-reference TE insertions, with RelocaTE2 showing the highest recall and precision in simulated data. We find that RelocaTE2, TEMP, TEMP2 and TEBreak provide consistent estimates of [Formula: see text]50 non-reference TE insertions per strain and that Ty2 has the highest number of non-reference TE insertions in a species-wide panel of [Formula: see text]1000 yeast genomes. Finally, we show that best-in-class predictors for yeast applied to resequencing data have sufficient resolution to reveal a dyad pattern of integration in nucleosome-bound regions upstream of yeast tRNA genes for Ty1, Ty2, and Ty4, allowing us to extend knowledge about fine-scale target preferences revealed previously for experimentally-induced Ty1 insertions to spontaneous insertions for other copia-superfamily retrotransposons in yeast.</p><p><strong>Conclusion: </strong>McClintock ( https://github.com/bergmanlab/mcclintock/ ) provides a user-friendly pipeline for the identification of TEs in short-read WGS data using multiple TE detectors, which should benefit researchers studying TE insertion variation in a wide range of different organisms. Application of the improved McClintock system to simulated and empirical yeast genome data reveals best-in-class methods and novel biological insights for one of the most widely-studied model eukaryotes and provides a paradigm for evaluating and selecting non-reference TE detectors in other species.</p>\",\"PeriodicalId\":18854,\"journal\":{\"name\":\"Mobile DNA\",\"volume\":\"14 1\",\"pages\":\"8\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2023-07-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10347736/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Mobile DNA\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13100-023-00296-4\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Mobile DNA","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13100-023-00296-4","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Reproducible evaluation of transposable element detectors with McClintock 2 guides accurate inference of Ty insertion patterns in yeast.
Background: Many computational methods have been developed to detect non-reference transposable element (TE) insertions using short-read whole genome sequencing data. The diversity and complexity of such methods often present challenges to new users seeking to reproducibly install, execute, or evaluate multiple TE insertion detectors.
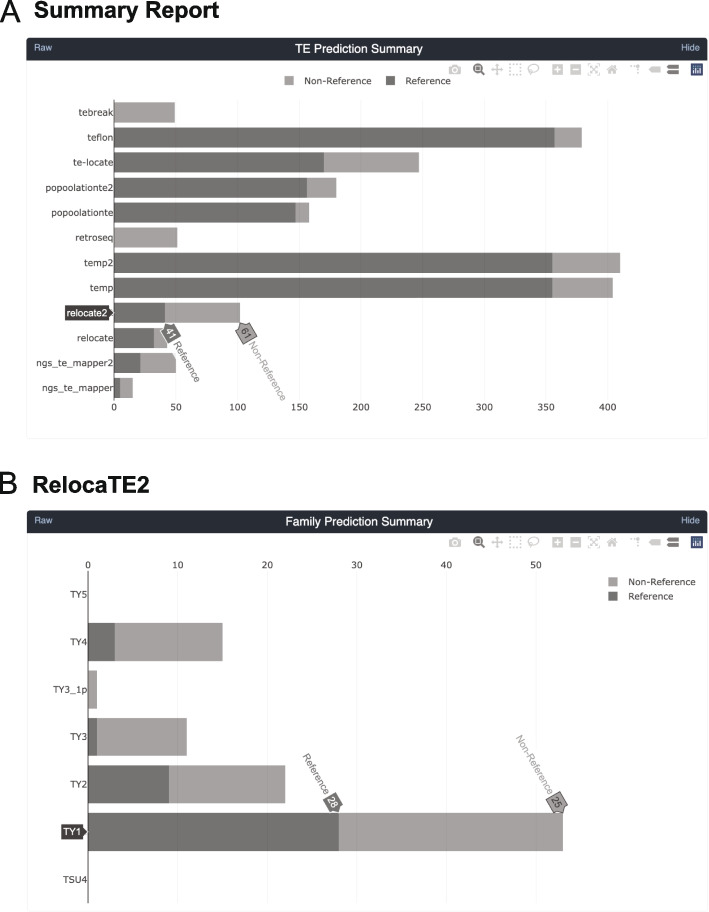
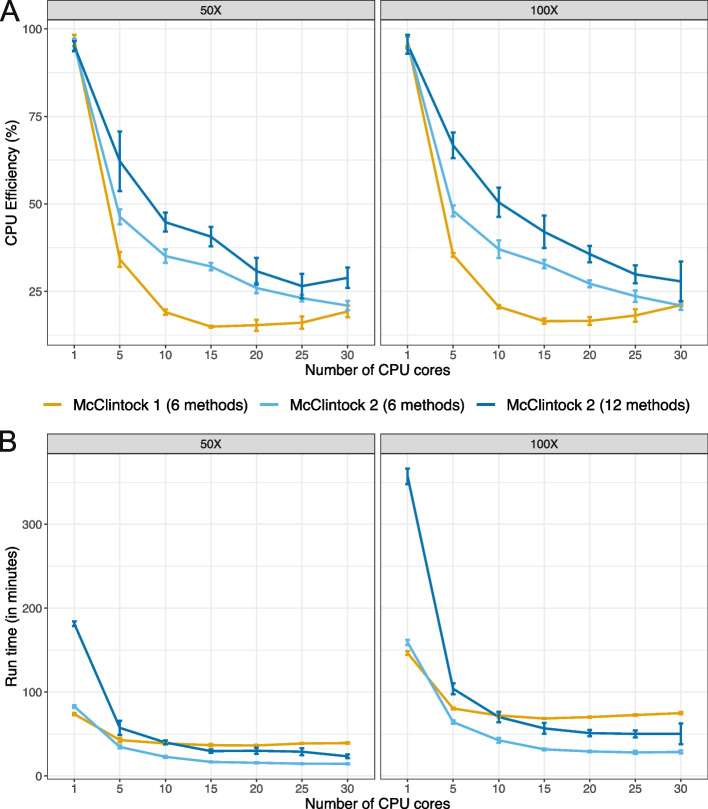
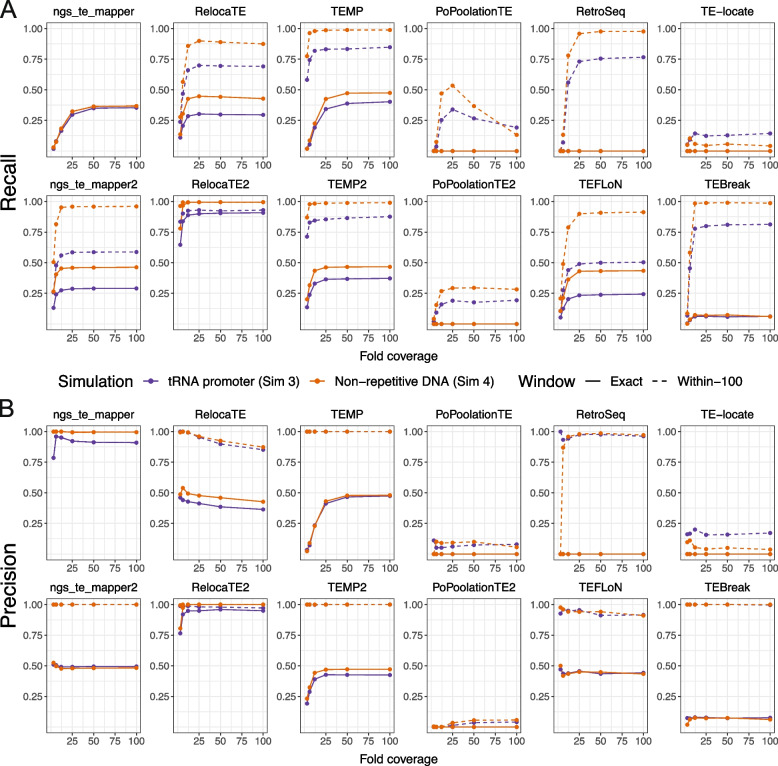
Results: We previously developed the McClintock meta-pipeline to facilitate the installation, execution, and evaluation of six first-generation short-read TE detectors. Here, we report a completely re-implemented version of McClintock written in Python using Snakemake and Conda that improves its installation, error handling, speed, stability, and extensibility. McClintock 2 now includes 12 short-read TE detectors, auxiliary pre-processing and analysis modules, interactive HTML reports, and a simulation framework to reproducibly evaluate the accuracy of component TE detectors. When applied to the model microbial eukaryote Saccharomyces cerevisiae, we find substantial variation in the ability of McClintock 2 components to identify the precise locations of non-reference TE insertions, with RelocaTE2 showing the highest recall and precision in simulated data. We find that RelocaTE2, TEMP, TEMP2 and TEBreak provide consistent estimates of [Formula: see text]50 non-reference TE insertions per strain and that Ty2 has the highest number of non-reference TE insertions in a species-wide panel of [Formula: see text]1000 yeast genomes. Finally, we show that best-in-class predictors for yeast applied to resequencing data have sufficient resolution to reveal a dyad pattern of integration in nucleosome-bound regions upstream of yeast tRNA genes for Ty1, Ty2, and Ty4, allowing us to extend knowledge about fine-scale target preferences revealed previously for experimentally-induced Ty1 insertions to spontaneous insertions for other copia-superfamily retrotransposons in yeast.
Conclusion: McClintock ( https://github.com/bergmanlab/mcclintock/ ) provides a user-friendly pipeline for the identification of TEs in short-read WGS data using multiple TE detectors, which should benefit researchers studying TE insertion variation in a wide range of different organisms. Application of the improved McClintock system to simulated and empirical yeast genome data reveals best-in-class methods and novel biological insights for one of the most widely-studied model eukaryotes and provides a paradigm for evaluating and selecting non-reference TE detectors in other species.
期刊介绍:
Mobile DNA is an online, peer-reviewed, open access journal that publishes articles providing novel insights into DNA rearrangements in all organisms, ranging from transposition and other types of recombination mechanisms to patterns and processes of mobile element and host genome evolution. In addition, the journal will consider articles on the utility of mobile genetic elements in biotechnological methods and protocols.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


