Bruno Bouça, Mariana Cascão, Pedro Fiúza, Sara Amaral, Paula Bogalho, José Silva-Nunes
{"title":"17- α羟化酶缺乏症的诊断是在46,XY核型患者的晚年进行的。","authors":"Bruno Bouça, Mariana Cascão, Pedro Fiúza, Sara Amaral, Paula Bogalho, José Silva-Nunes","doi":"10.1530/EDM-22-0338","DOIUrl":null,"url":null,"abstract":"<p><strong>Summary: </strong>17-Alpha-hydroxylase deficiency (17OHD) is a rare autosomal recessive disease, representing 1% of cases of congenital adrenal hyperplasia. A 44-year-old female presented to the emergency department complaining of generalized asthenia and polyarthralgia for about 2 weeks. On examination, she was hypertensive (174/100 mmHg), and laboratory results revealed hypokalemia and hypocortisolism. She had an uncharacteristic morphotype, BMI of 16.7 kg/m2, cutaneous hyperpigmentation, and Tanner stage M1P1, with normal female external genitalia. She reported to have primary amenorrhea. Further analytical evaluations of her hormone levels were performed CT scan revealed adrenal bilateral hyperplasia and absence of female internal genitalia. A nodular lesion was observed in the left inguinal canal with 25 × 10 mm, compatible with a testicular remnant. Genetic analysis identified the c.3G>A p.(Met1?) variant in homozygosity in the CYP17A1 gene, classified as pathogenic, confirming the diagnosis of 17OHD. Karyotype analysis was compatible with 46,XY. The association of severe hypokalemia, hypertension, hypocortisolism, and oligo/amenorrhea and the absence of secondary sexual characteristics favored the diagnosis of 17OHD, confirmed by genetic testing. As in other published clinical cases, diagnosis outside pediatric age is not rare and should be considered when severe hypokalemia occurs in hypertensive adults with a lack of secondary sexual characteristics.</p><p><strong>Learning points: </strong>The association of severe hypokalemia, hypertension, hypocortisolism, and oligo/amenorrhea and the absence of secondary sexual characteristics favor the diagnosis of 17-alpha-hydroxylase deficiency (17OHD). Diagnosis outside pediatric age is not rare. 17OHD should be considered when severe hypokalemia occurs in hypertensive adults with a lack of secondary sexual characteristics.</p>","PeriodicalId":37467,"journal":{"name":"Endocrinology, Diabetes and Metabolism Case Reports","volume":null,"pages":null},"PeriodicalIF":0.7000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10337863/pdf/","citationCount":"0","resultStr":"{\"title\":\"Diagnosis of 17-alpha hydroxylase deficiency performed late in life in a patient with a 46,XY karyotype.\",\"authors\":\"Bruno Bouça, Mariana Cascão, Pedro Fiúza, Sara Amaral, Paula Bogalho, José Silva-Nunes\",\"doi\":\"10.1530/EDM-22-0338\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Summary: </strong>17-Alpha-hydroxylase deficiency (17OHD) is a rare autosomal recessive disease, representing 1% of cases of congenital adrenal hyperplasia. A 44-year-old female presented to the emergency department complaining of generalized asthenia and polyarthralgia for about 2 weeks. On examination, she was hypertensive (174/100 mmHg), and laboratory results revealed hypokalemia and hypocortisolism. She had an uncharacteristic morphotype, BMI of 16.7 kg/m2, cutaneous hyperpigmentation, and Tanner stage M1P1, with normal female external genitalia. She reported to have primary amenorrhea. Further analytical evaluations of her hormone levels were performed CT scan revealed adrenal bilateral hyperplasia and absence of female internal genitalia. A nodular lesion was observed in the left inguinal canal with 25 × 10 mm, compatible with a testicular remnant. Genetic analysis identified the c.3G>A p.(Met1?) variant in homozygosity in the CYP17A1 gene, classified as pathogenic, confirming the diagnosis of 17OHD. Karyotype analysis was compatible with 46,XY. The association of severe hypokalemia, hypertension, hypocortisolism, and oligo/amenorrhea and the absence of secondary sexual characteristics favored the diagnosis of 17OHD, confirmed by genetic testing. As in other published clinical cases, diagnosis outside pediatric age is not rare and should be considered when severe hypokalemia occurs in hypertensive adults with a lack of secondary sexual characteristics.</p><p><strong>Learning points: </strong>The association of severe hypokalemia, hypertension, hypocortisolism, and oligo/amenorrhea and the absence of secondary sexual characteristics favor the diagnosis of 17-alpha-hydroxylase deficiency (17OHD). Diagnosis outside pediatric age is not rare. 17OHD should be considered when severe hypokalemia occurs in hypertensive adults with a lack of secondary sexual characteristics.</p>\",\"PeriodicalId\":37467,\"journal\":{\"name\":\"Endocrinology, Diabetes and Metabolism Case Reports\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":0.7000,\"publicationDate\":\"2023-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10337863/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Endocrinology, Diabetes and Metabolism Case Reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1530/EDM-22-0338\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Endocrinology, Diabetes and Metabolism Case Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1530/EDM-22-0338","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Diagnosis of 17-alpha hydroxylase deficiency performed late in life in a patient with a 46,XY karyotype.
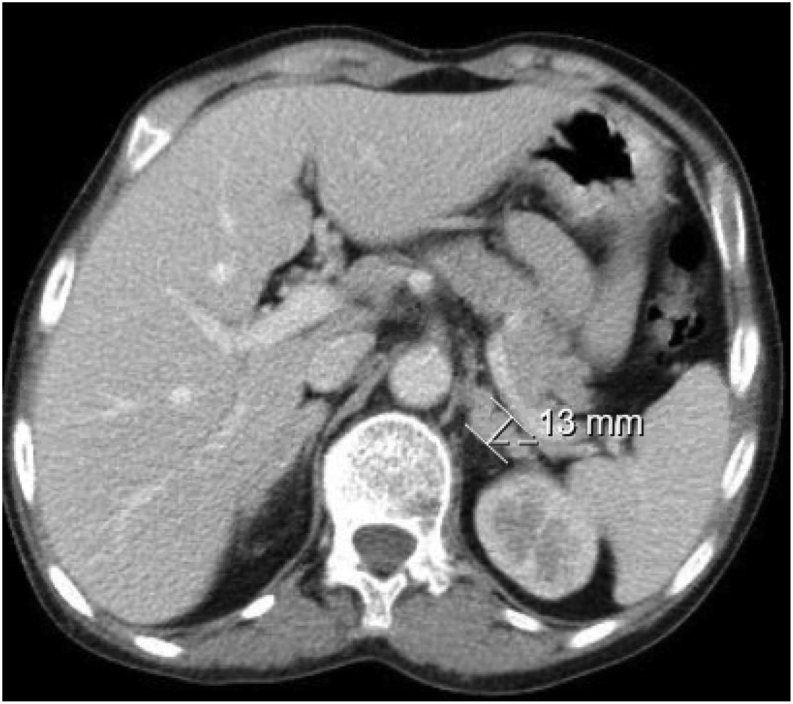
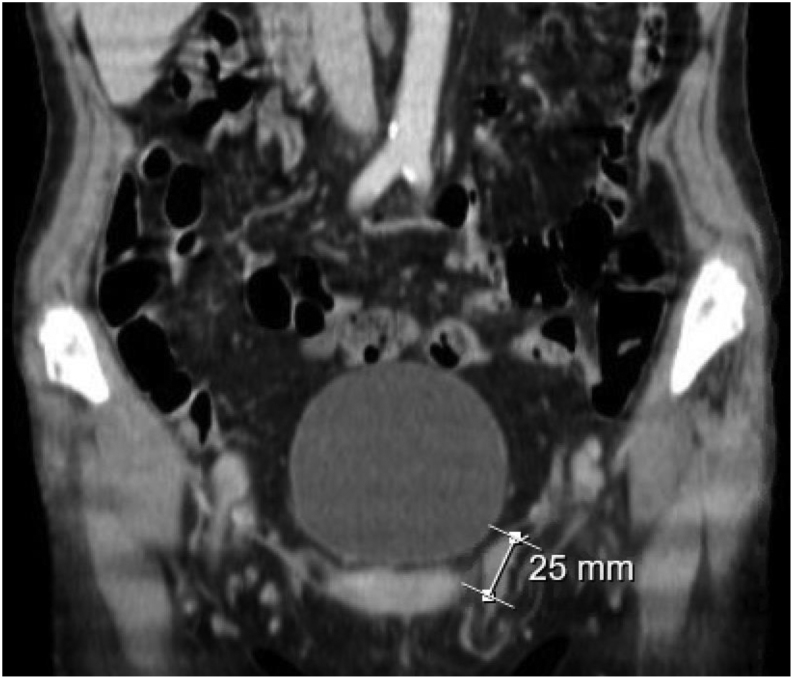
Summary: 17-Alpha-hydroxylase deficiency (17OHD) is a rare autosomal recessive disease, representing 1% of cases of congenital adrenal hyperplasia. A 44-year-old female presented to the emergency department complaining of generalized asthenia and polyarthralgia for about 2 weeks. On examination, she was hypertensive (174/100 mmHg), and laboratory results revealed hypokalemia and hypocortisolism. She had an uncharacteristic morphotype, BMI of 16.7 kg/m2, cutaneous hyperpigmentation, and Tanner stage M1P1, with normal female external genitalia. She reported to have primary amenorrhea. Further analytical evaluations of her hormone levels were performed CT scan revealed adrenal bilateral hyperplasia and absence of female internal genitalia. A nodular lesion was observed in the left inguinal canal with 25 × 10 mm, compatible with a testicular remnant. Genetic analysis identified the c.3G>A p.(Met1?) variant in homozygosity in the CYP17A1 gene, classified as pathogenic, confirming the diagnosis of 17OHD. Karyotype analysis was compatible with 46,XY. The association of severe hypokalemia, hypertension, hypocortisolism, and oligo/amenorrhea and the absence of secondary sexual characteristics favored the diagnosis of 17OHD, confirmed by genetic testing. As in other published clinical cases, diagnosis outside pediatric age is not rare and should be considered when severe hypokalemia occurs in hypertensive adults with a lack of secondary sexual characteristics.
Learning points: The association of severe hypokalemia, hypertension, hypocortisolism, and oligo/amenorrhea and the absence of secondary sexual characteristics favor the diagnosis of 17-alpha-hydroxylase deficiency (17OHD). Diagnosis outside pediatric age is not rare. 17OHD should be considered when severe hypokalemia occurs in hypertensive adults with a lack of secondary sexual characteristics.
期刊介绍:
Endocrinology, Diabetes & Metabolism Case Reports publishes case reports on common and rare conditions in all areas of clinical endocrinology, diabetes and metabolism. Articles should include clear learning points which readers can use to inform medical education or clinical practice. The types of cases of interest to Endocrinology, Diabetes & Metabolism Case Reports include: -Insight into disease pathogenesis or mechanism of therapy - Novel diagnostic procedure - Novel treatment - Unique/unexpected symptoms or presentations of a disease - New disease or syndrome: presentations/diagnosis/management - Unusual effects of medical treatment - Error in diagnosis/pitfalls and caveats


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


