{"title":"青少年息肉病综合征中BMPR1a致病变异的基因型-表型相关性","authors":"M E Papadopulos, J P Plazzer, F A Macrae","doi":"10.1186/s13053-023-00255-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Juvenile Polyposis Syndrome (JPS) is an autosomal dominant condition with hamartomatous polyps in the gastrointestinal tract, associated with an increased risk of gastrointestinal malignancy. Disease causing variants (DCVs) in BMPR1a or SMAD4 account for 45-60% of JPS cases, with BMPR1a DCVs accounting for 17-38% of JPS cases. Within those with either a BMPR1a or SMAD4 DCV, there is phenotypic variability in location of polyps, risk of malignancy and extra-intestinal manifestations with limited published reports of gene-phenotype association or genotype-phenotype correlation. We aimed to identify any gene-phenotype association or genotype-phenotype correlation in BMPR1a to inform surveillance recommendations, and gene-specific modification to the ACMG classification of pathogenicity of DCVs.</p><p><strong>Methods: </strong>A literature search was performed through EMBASE, MEDLINE and PubMed. Studies that were included explored BMPR1a DCV-related JPS or contiguous deletion of PTEN and BMPR1a. Data was also drawn from the BMPR1a specific databases on LOVD and ClinVar.</p><p><strong>Results: </strong>There were 211 DCVs in BMPR1a identified, 82 from patients with JPS in the literature, and 17 from LOVD and 112 from ClinVar classified as pathogenic or likely pathogenic. These included missense, nonsense and frameshift variants and large deletions, occurring across all functional domains of the gene. Unlike in SMAD4 carriers, gastric polyposis and malignancy were not identified in our review in BMPR1a carriers, but colonic polyposis and malignancy occurred in carriers of either BMPR1a or SMAD4 DCVs. Those with contiguous deletion of PTEN and BMPR1a can present with JPS of infancy, with a severe phenotype of GI bleeding, diarrhoea, exudative enteropathy and rectal prolapse. No specific BMPR1a genotype-phenotype correlation could be ascertained including by variant type or functional domain.</p><p><strong>Conclusion: </strong>Phenotypic characteristics cannot be used to inform variant location in BMPR1a. However, the phenotypic characteristics of BMPR1a DCV carriers, being almost exclusively related to the colon and rectum, can assist in pathogenicity assessment of BMPR1a variants. Given these findings, we propose that carriers of BMPR1a DCVs should only require surveillance for colorectal polyps and malignancy, and that surveillance for gastric polyps and malignancy may be unnecessary. However variant location within BMPR1a does not support differential surveillance recommendations.</p>","PeriodicalId":55058,"journal":{"name":"Hereditary Cancer in Clinical Practice","volume":"21 1","pages":"12"},"PeriodicalIF":2.4000,"publicationDate":"2023-07-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10316536/pdf/","citationCount":"2","resultStr":"{\"title\":\"Genotype-phenotype correlation of BMPR1a disease causing variants in juvenile polyposis syndrome.\",\"authors\":\"M E Papadopulos, J P Plazzer, F A Macrae\",\"doi\":\"10.1186/s13053-023-00255-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Juvenile Polyposis Syndrome (JPS) is an autosomal dominant condition with hamartomatous polyps in the gastrointestinal tract, associated with an increased risk of gastrointestinal malignancy. Disease causing variants (DCVs) in BMPR1a or SMAD4 account for 45-60% of JPS cases, with BMPR1a DCVs accounting for 17-38% of JPS cases. Within those with either a BMPR1a or SMAD4 DCV, there is phenotypic variability in location of polyps, risk of malignancy and extra-intestinal manifestations with limited published reports of gene-phenotype association or genotype-phenotype correlation. We aimed to identify any gene-phenotype association or genotype-phenotype correlation in BMPR1a to inform surveillance recommendations, and gene-specific modification to the ACMG classification of pathogenicity of DCVs.</p><p><strong>Methods: </strong>A literature search was performed through EMBASE, MEDLINE and PubMed. Studies that were included explored BMPR1a DCV-related JPS or contiguous deletion of PTEN and BMPR1a. Data was also drawn from the BMPR1a specific databases on LOVD and ClinVar.</p><p><strong>Results: </strong>There were 211 DCVs in BMPR1a identified, 82 from patients with JPS in the literature, and 17 from LOVD and 112 from ClinVar classified as pathogenic or likely pathogenic. These included missense, nonsense and frameshift variants and large deletions, occurring across all functional domains of the gene. Unlike in SMAD4 carriers, gastric polyposis and malignancy were not identified in our review in BMPR1a carriers, but colonic polyposis and malignancy occurred in carriers of either BMPR1a or SMAD4 DCVs. Those with contiguous deletion of PTEN and BMPR1a can present with JPS of infancy, with a severe phenotype of GI bleeding, diarrhoea, exudative enteropathy and rectal prolapse. No specific BMPR1a genotype-phenotype correlation could be ascertained including by variant type or functional domain.</p><p><strong>Conclusion: </strong>Phenotypic characteristics cannot be used to inform variant location in BMPR1a. However, the phenotypic characteristics of BMPR1a DCV carriers, being almost exclusively related to the colon and rectum, can assist in pathogenicity assessment of BMPR1a variants. Given these findings, we propose that carriers of BMPR1a DCVs should only require surveillance for colorectal polyps and malignancy, and that surveillance for gastric polyps and malignancy may be unnecessary. However variant location within BMPR1a does not support differential surveillance recommendations.</p>\",\"PeriodicalId\":55058,\"journal\":{\"name\":\"Hereditary Cancer in Clinical Practice\",\"volume\":\"21 1\",\"pages\":\"12\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2023-07-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10316536/pdf/\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Hereditary Cancer in Clinical Practice\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13053-023-00255-3\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"ONCOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hereditary Cancer in Clinical Practice","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13053-023-00255-3","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ONCOLOGY","Score":null,"Total":0}
Genotype-phenotype correlation of BMPR1a disease causing variants in juvenile polyposis syndrome.
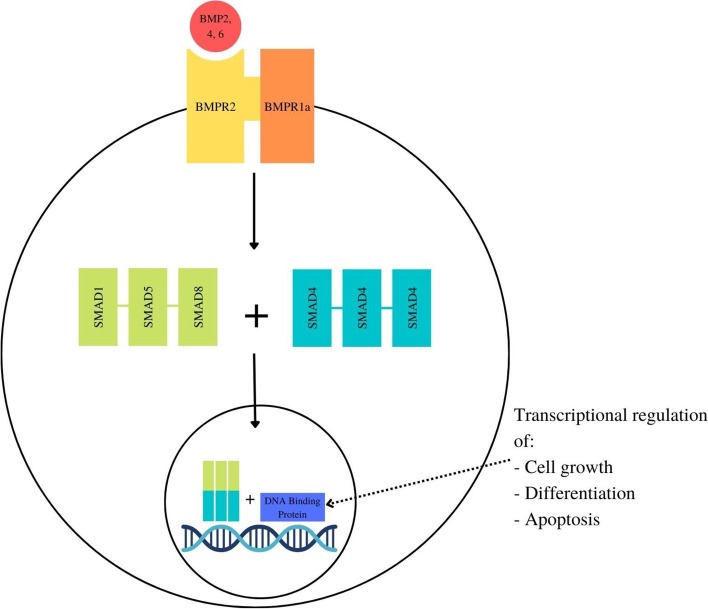
Background: Juvenile Polyposis Syndrome (JPS) is an autosomal dominant condition with hamartomatous polyps in the gastrointestinal tract, associated with an increased risk of gastrointestinal malignancy. Disease causing variants (DCVs) in BMPR1a or SMAD4 account for 45-60% of JPS cases, with BMPR1a DCVs accounting for 17-38% of JPS cases. Within those with either a BMPR1a or SMAD4 DCV, there is phenotypic variability in location of polyps, risk of malignancy and extra-intestinal manifestations with limited published reports of gene-phenotype association or genotype-phenotype correlation. We aimed to identify any gene-phenotype association or genotype-phenotype correlation in BMPR1a to inform surveillance recommendations, and gene-specific modification to the ACMG classification of pathogenicity of DCVs.
Methods: A literature search was performed through EMBASE, MEDLINE and PubMed. Studies that were included explored BMPR1a DCV-related JPS or contiguous deletion of PTEN and BMPR1a. Data was also drawn from the BMPR1a specific databases on LOVD and ClinVar.
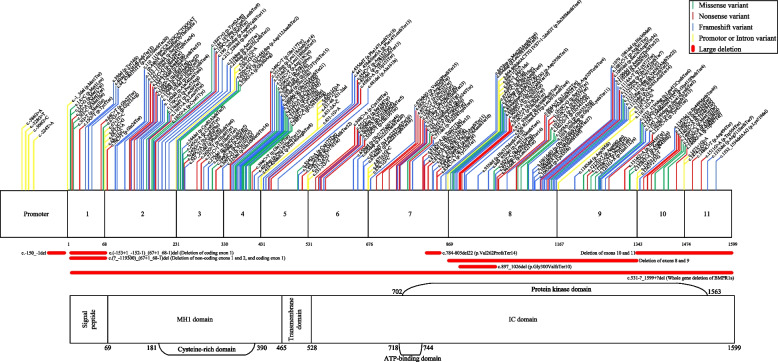
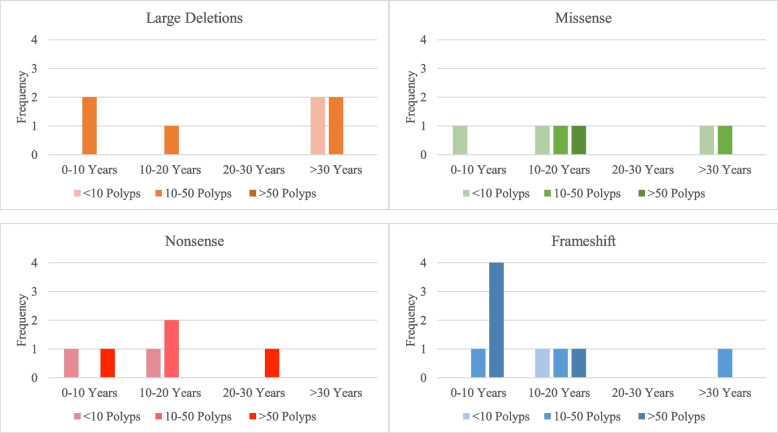
Results: There were 211 DCVs in BMPR1a identified, 82 from patients with JPS in the literature, and 17 from LOVD and 112 from ClinVar classified as pathogenic or likely pathogenic. These included missense, nonsense and frameshift variants and large deletions, occurring across all functional domains of the gene. Unlike in SMAD4 carriers, gastric polyposis and malignancy were not identified in our review in BMPR1a carriers, but colonic polyposis and malignancy occurred in carriers of either BMPR1a or SMAD4 DCVs. Those with contiguous deletion of PTEN and BMPR1a can present with JPS of infancy, with a severe phenotype of GI bleeding, diarrhoea, exudative enteropathy and rectal prolapse. No specific BMPR1a genotype-phenotype correlation could be ascertained including by variant type or functional domain.
Conclusion: Phenotypic characteristics cannot be used to inform variant location in BMPR1a. However, the phenotypic characteristics of BMPR1a DCV carriers, being almost exclusively related to the colon and rectum, can assist in pathogenicity assessment of BMPR1a variants. Given these findings, we propose that carriers of BMPR1a DCVs should only require surveillance for colorectal polyps and malignancy, and that surveillance for gastric polyps and malignancy may be unnecessary. However variant location within BMPR1a does not support differential surveillance recommendations.
期刊介绍:
Hereditary Cancer in Clinical Practice is an open access journal that publishes articles of interest for the cancer genetics community and serves as a discussion forum for the development appropriate healthcare strategies.
Cancer genetics encompasses a wide variety of disciplines and knowledge in the field is rapidly growing, especially as the amount of information linking genetic differences to inherited cancer predispositions continues expanding. With the increased knowledge of genetic variability and how this relates to cancer risk there is a growing demand not only to disseminate this information into clinical practice but also to enable competent debate concerning how such information is managed and what it implies for patient care.
Topics covered by the journal include but are not limited to:
Original research articles on any aspect of inherited predispositions to cancer.
Reviews of inherited cancer predispositions.
Application of molecular and cytogenetic analysis to clinical decision making.
Clinical aspects of the management of hereditary cancers.
Genetic counselling issues associated with cancer genetics.
The role of registries in improving health care of patients with an inherited predisposition to cancer.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


