Pavel V Afonine, Paul D Adams, Alexandre G Urzhumtsev
{"title":"多成分晶体的高效结构因子建模。","authors":"Pavel V Afonine, Paul D Adams, Alexandre G Urzhumtsev","doi":"10.1107/S205327332300356X","DOIUrl":null,"url":null,"abstract":"<p><p>Diffraction intensities from a crystallographic experiment include contributions from the entire unit cell of the crystal: the macromolecule, the solvent around it and eventually other compounds. These contributions cannot typically be well described by an atomic model alone, i.e. using point scatterers. Indeed, entities such as disordered (bulk) solvent, semi-ordered solvent (e.g. lipid belts in membrane proteins, ligands, ion channels) and disordered polymer loops require other types of modeling than a collection of individual atoms. This results in the model structure factors containing multiple contributions. Most macromolecular applications assume two-component structure factors: one component arising from the atomic model and the second one describing the bulk solvent. A more accurate and detailed modeling of the disordered regions of the crystal will naturally require more than two components in the structure factors, which presents algorithmic and computational challenges. Here an efficient solution of this problem is proposed. All algorithms described in this work have been implemented in the computational crystallography toolbox (CCTBX) and are also available within Phenix software. These algorithms are rather general and do not use any assumptions about molecule type or size nor about those of its components.</p>","PeriodicalId":106,"journal":{"name":"Acta Crystallographica Section A: Foundations and Advances","volume":"79 Pt 4","pages":"345-352"},"PeriodicalIF":1.8000,"publicationDate":"2023-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10317137/pdf/","citationCount":"0","resultStr":"{\"title\":\"Efficient structure-factor modeling for crystals with multiple components.\",\"authors\":\"Pavel V Afonine, Paul D Adams, Alexandre G Urzhumtsev\",\"doi\":\"10.1107/S205327332300356X\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Diffraction intensities from a crystallographic experiment include contributions from the entire unit cell of the crystal: the macromolecule, the solvent around it and eventually other compounds. These contributions cannot typically be well described by an atomic model alone, i.e. using point scatterers. Indeed, entities such as disordered (bulk) solvent, semi-ordered solvent (e.g. lipid belts in membrane proteins, ligands, ion channels) and disordered polymer loops require other types of modeling than a collection of individual atoms. This results in the model structure factors containing multiple contributions. Most macromolecular applications assume two-component structure factors: one component arising from the atomic model and the second one describing the bulk solvent. A more accurate and detailed modeling of the disordered regions of the crystal will naturally require more than two components in the structure factors, which presents algorithmic and computational challenges. Here an efficient solution of this problem is proposed. All algorithms described in this work have been implemented in the computational crystallography toolbox (CCTBX) and are also available within Phenix software. These algorithms are rather general and do not use any assumptions about molecule type or size nor about those of its components.</p>\",\"PeriodicalId\":106,\"journal\":{\"name\":\"Acta Crystallographica Section A: Foundations and Advances\",\"volume\":\"79 Pt 4\",\"pages\":\"345-352\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2023-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10317137/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Crystallographica Section A: Foundations and Advances\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1107/S205327332300356X\",\"RegionNum\":4,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/6/20 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Crystallographica Section A: Foundations and Advances","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1107/S205327332300356X","RegionNum":4,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/6/20 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Efficient structure-factor modeling for crystals with multiple components.
Diffraction intensities from a crystallographic experiment include contributions from the entire unit cell of the crystal: the macromolecule, the solvent around it and eventually other compounds. These contributions cannot typically be well described by an atomic model alone, i.e. using point scatterers. Indeed, entities such as disordered (bulk) solvent, semi-ordered solvent (e.g. lipid belts in membrane proteins, ligands, ion channels) and disordered polymer loops require other types of modeling than a collection of individual atoms. This results in the model structure factors containing multiple contributions. Most macromolecular applications assume two-component structure factors: one component arising from the atomic model and the second one describing the bulk solvent. A more accurate and detailed modeling of the disordered regions of the crystal will naturally require more than two components in the structure factors, which presents algorithmic and computational challenges. Here an efficient solution of this problem is proposed. All algorithms described in this work have been implemented in the computational crystallography toolbox (CCTBX) and are also available within Phenix software. These algorithms are rather general and do not use any assumptions about molecule type or size nor about those of its components.
期刊介绍:
Acta Crystallographica Section A: Foundations and Advances publishes articles reporting advances in the theory and practice of all areas of crystallography in the broadest sense. As well as traditional crystallography, this includes nanocrystals, metacrystals, amorphous materials, quasicrystals, synchrotron and XFEL studies, coherent scattering, diffraction imaging, time-resolved studies and the structure of strain and defects in materials.
The journal has two parts, a rapid-publication Advances section and the traditional Foundations section. Articles for the Advances section are of particularly high value and impact. They receive expedited treatment and may be highlighted by an accompanying scientific commentary article and a press release. Further details are given in the November 2013 Editorial.
The central themes of the journal are, on the one hand, experimental and theoretical studies of the properties and arrangements of atoms, ions and molecules in condensed matter, periodic, quasiperiodic or amorphous, ideal or real, and, on the other, the theoretical and experimental aspects of the various methods to determine these properties and arrangements.
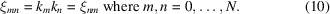
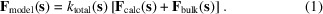

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


