{"title":"Lynch综合征相关脊索瘤具有高肿瘤突变负担和对免疫检查点抑制剂的显著反应。","authors":"Naoki Shinojima, Kazutaka Ozono, Haruaki Yamamoto, Sakiko Abe, Rumi Sasaki, Yusuke Tomita, Azusa Kai, Ryosuke Mori, Takahiro Yamamoto, Ken Uekawa, Hirotaka Matsui, Kisato Nosaka, Hiroaki Matsuzaki, Yoshihiro Komohara, Yoshiki Mikami, Akitake Mukasa","doi":"10.1007/s10014-023-00461-w","DOIUrl":null,"url":null,"abstract":"<p><p>Chordoma is a rare malignant bone tumor arising from notochordal tissue. Conventional treatments, such as radical resection and high-dose irradiation, frequently fail to control the tumor, resulting in recurrence and re-growth. In this study, genetic analysis of the tumor in a 72-year-old male patient with refractory conventional chordoma of the skull base revealed a high tumor mutational burden (TMB) and mutations in the MSH6 and MLH1 genes, which are found in Lynch syndrome. The patient and his family had a dense cancer history, and subsequent germline genetic testing revealed Lynch syndrome. This is the first report of a chordoma that has been genetically proven to be Lynch syndrome. Chordomas usually have low TMB; however, this is an unusual case, because the TMB was high, and immune checkpoint inhibitors effectively controlled the tumor. This case provides a basis for determining the indications for immunotherapy of chordoma based on the genetic analysis. Therefore, further extensive genetic analysis in the future will help to stratify the treatment of chordoma.</p>","PeriodicalId":9226,"journal":{"name":"Brain Tumor Pathology","volume":"40 3","pages":"185-190"},"PeriodicalIF":3.0000,"publicationDate":"2023-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10314830/pdf/","citationCount":"0","resultStr":"{\"title\":\"Lynch syndrome-associated chordoma with high tumor mutational burden and significant response to immune checkpoint inhibitors.\",\"authors\":\"Naoki Shinojima, Kazutaka Ozono, Haruaki Yamamoto, Sakiko Abe, Rumi Sasaki, Yusuke Tomita, Azusa Kai, Ryosuke Mori, Takahiro Yamamoto, Ken Uekawa, Hirotaka Matsui, Kisato Nosaka, Hiroaki Matsuzaki, Yoshihiro Komohara, Yoshiki Mikami, Akitake Mukasa\",\"doi\":\"10.1007/s10014-023-00461-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Chordoma is a rare malignant bone tumor arising from notochordal tissue. Conventional treatments, such as radical resection and high-dose irradiation, frequently fail to control the tumor, resulting in recurrence and re-growth. In this study, genetic analysis of the tumor in a 72-year-old male patient with refractory conventional chordoma of the skull base revealed a high tumor mutational burden (TMB) and mutations in the MSH6 and MLH1 genes, which are found in Lynch syndrome. The patient and his family had a dense cancer history, and subsequent germline genetic testing revealed Lynch syndrome. This is the first report of a chordoma that has been genetically proven to be Lynch syndrome. Chordomas usually have low TMB; however, this is an unusual case, because the TMB was high, and immune checkpoint inhibitors effectively controlled the tumor. This case provides a basis for determining the indications for immunotherapy of chordoma based on the genetic analysis. Therefore, further extensive genetic analysis in the future will help to stratify the treatment of chordoma.</p>\",\"PeriodicalId\":9226,\"journal\":{\"name\":\"Brain Tumor Pathology\",\"volume\":\"40 3\",\"pages\":\"185-190\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2023-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10314830/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Brain Tumor Pathology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10014-023-00461-w\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain Tumor Pathology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10014-023-00461-w","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Lynch syndrome-associated chordoma with high tumor mutational burden and significant response to immune checkpoint inhibitors.
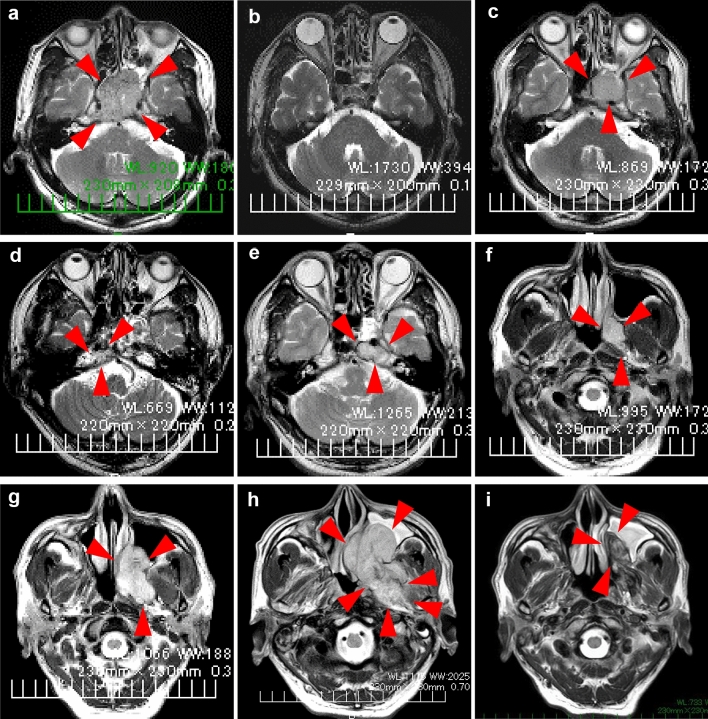
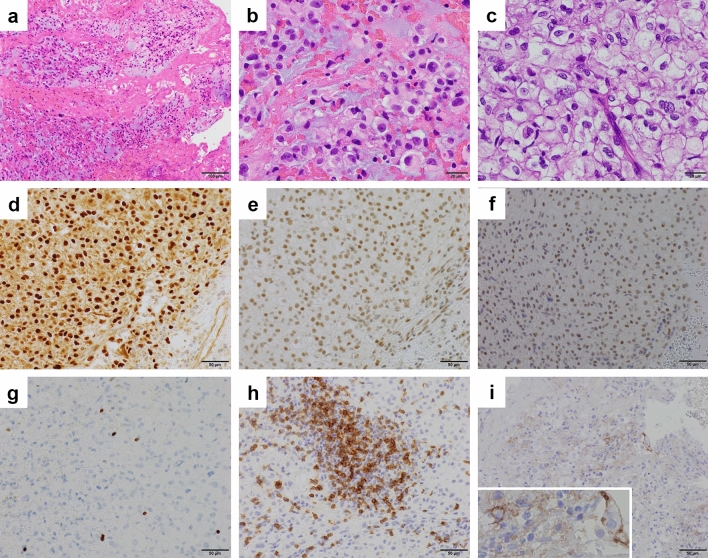
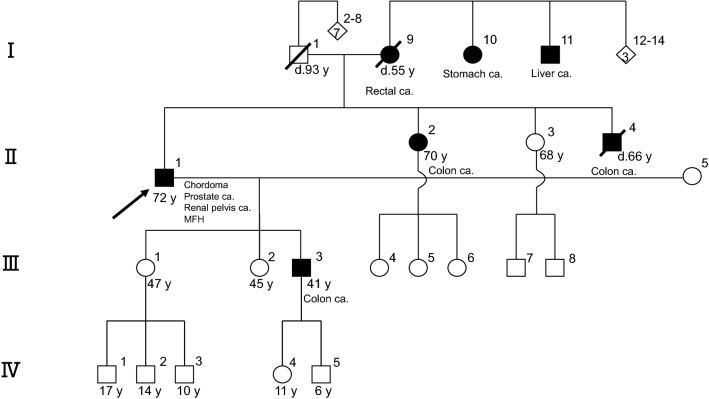
Chordoma is a rare malignant bone tumor arising from notochordal tissue. Conventional treatments, such as radical resection and high-dose irradiation, frequently fail to control the tumor, resulting in recurrence and re-growth. In this study, genetic analysis of the tumor in a 72-year-old male patient with refractory conventional chordoma of the skull base revealed a high tumor mutational burden (TMB) and mutations in the MSH6 and MLH1 genes, which are found in Lynch syndrome. The patient and his family had a dense cancer history, and subsequent germline genetic testing revealed Lynch syndrome. This is the first report of a chordoma that has been genetically proven to be Lynch syndrome. Chordomas usually have low TMB; however, this is an unusual case, because the TMB was high, and immune checkpoint inhibitors effectively controlled the tumor. This case provides a basis for determining the indications for immunotherapy of chordoma based on the genetic analysis. Therefore, further extensive genetic analysis in the future will help to stratify the treatment of chordoma.
期刊介绍:
Brain Tumor Pathology is the official journal of the Japan Society of Brain Tumor Pathology. This international journal documents the latest research and topical debate in all clinical and experimental fields relating to brain tumors, especially brain tumor pathology. The journal has been published since 1983 and has been recognized worldwide as a unique journal of high quality. The journal welcomes the submission of manuscripts from any country. Membership in the society is not a prerequisite for submission. The journal publishes original articles, case reports, rapid short communications, instructional lectures, review articles, letters to the editor, and topics.Review articles and Topics may be recommended at the annual meeting of the Japan Society of Brain Tumor Pathology. All contributions should be aimed at promoting international scientific collaboration.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


