通过密码子优化的 cDNA 转基因增强人类生存运动神经元 1 基因在体外和体内的表达。
IF 4.5
3区 医学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
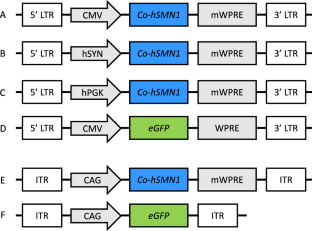
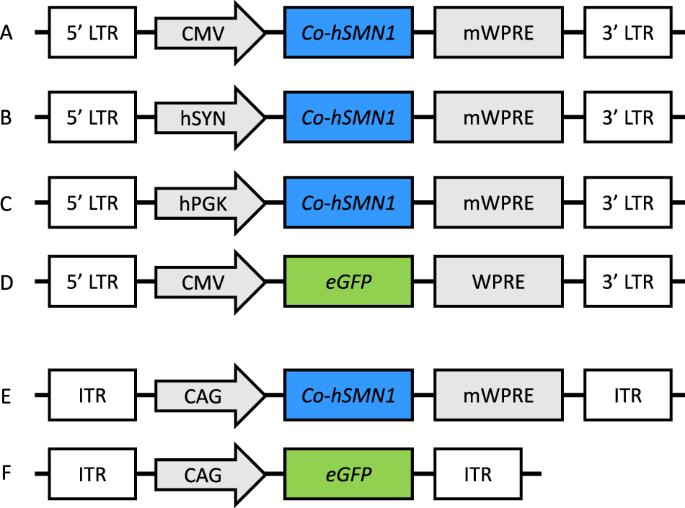
脊髓性肌萎缩症(SMA)是一种以腹侧运动神经元变性为主要特征的神经肌肉疾病。存活运动神经元(SMN)1基因突变会导致SMA,而替换有问题的SMN1拷贝的基因添加策略是一种治疗选择。我们开发了一种新颖的、经过密码子优化的 hSMN1 转基因,并生产了带有巨细胞病毒(CMV)、人突触素(hSYN)或人磷酸甘油激酶(hPGK)启动子的整合良好和整合不良慢病毒载体,以确定最佳的表达盒配置。整合、CMV 驱动和密码子优化的 hSMN1 慢病毒载体在体外产生的功能性 SMN 蛋白最高。整合缺陷的慢病毒载体也能显著表达优化的转基因,预计比整合载体更安全。慢病毒载体在培养过程中会导致 DNA 损伤反应的激活,特别是使磷酸化共济失调毛细血管扩张症突变体(pATM)和 γH2AX 的水平升高,但优化的 hSMN1 转基因显示出一定的保护作用。新生儿期将编码优化转基因的腺相关病毒载体(AAV9)输送到Smn2B/-SMA模型小鼠体内,可显著提高肝脏和脊髓中的SMN蛋白水平。这项研究表明,一种新的经过密码子优化的 hSMN1 转基因有可能成为 SMA 的一种治疗策略。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Enhanced expression of the human Survival motor neuron 1 gene from a codon-optimised cDNA transgene in vitro and in vivo
Spinal muscular atrophy (SMA) is a neuromuscular disease particularly characterised by degeneration of ventral motor neurons. Survival motor neuron (SMN) 1 gene mutations cause SMA, and gene addition strategies to replace the faulty SMN1 copy are a therapeutic option. We have developed a novel, codon-optimised hSMN1 transgene and produced integration-proficient and integration-deficient lentiviral vectors with cytomegalovirus (CMV), human synapsin (hSYN) or human phosphoglycerate kinase (hPGK) promoters to determine the optimal expression cassette configuration. Integrating, CMV-driven and codon-optimised hSMN1 lentiviral vectors resulted in the highest production of functional SMN protein in vitro. Integration-deficient lentiviral vectors also led to significant expression of the optimised transgene and are expected to be safer than integrating vectors. Lentiviral delivery in culture led to activation of the DNA damage response, in particular elevating levels of phosphorylated ataxia telangiectasia mutated (pATM) and γH2AX, but the optimised hSMN1 transgene showed some protective effects. Neonatal delivery of adeno-associated viral vector (AAV9) vector encoding the optimised transgene to the Smn2B/− mouse model of SMA resulted in a significant increase of SMN protein levels in liver and spinal cord. This work shows the potential of a novel codon-optimised hSMN1 transgene as a therapeutic strategy for SMA.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Gene Therapy
医学-生化与分子生物学
CiteScore
9.70
自引率
2.00%
发文量
67
审稿时长
4-8 weeks
期刊介绍:
Gene Therapy covers both the research and clinical applications of novel therapeutic techniques based on a genetic component. Over the last few decades, significant advances in technologies ranging from identifying novel genetic targets that cause disease through to clinical studies, which show therapeutic benefit, have elevated this multidisciplinary field to the forefront of modern medicine.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


