Foeaz Ahmed, Md Nazmul Islam Bappy, Md Shariful Islam
{"title":"麻疯树中保守mirna及其靶点的鉴定:一种计算机方法。","authors":"Foeaz Ahmed, Md Nazmul Islam Bappy, Md Shariful Islam","doi":"10.1186/s43141-023-00495-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>MicroRNAs (miRNAs) are small endogenous RNAs with an approximate length of 18-22 nucleotides and involved in the regulation of gene expression in transcriptional or post-transcriptional levels. They were found to be associated with leaf morphogenesis, flowering time, vegetative phase change, and response to environmental cues in plants, where they act as a critical regulatory factor. The nature of high conservancy of plant miRNAs within the plant species made it possible to detect the conserved miRNAs by computational approaches. Expressed Sequence Tags (EST) based comparative genomic approaches provide advantages over wet lab approaches as it is convenient, easy to carry out and less time consuming. EST-based in silico approach can unravel new conserved miRNAs in plants, even when the complete genome sequence is not available.</p><p><strong>Results: </strong>To identify the novel miRNAs, a total of 46,865 ESTs from Jatropha curcas were searched for homology to all available 6746 mature miRNAs of plant eudicotyledons. Finally, we ended up with 12 novel miRNAs in Jatropha that range from 18 to 19 nucleotides where their respective precursor miRNAs had 54.11-71.76% (A + U) content. The putative miRNAs belong to 12 individual miRNA family and most of them have higher (A + U) content ranging from 47.36 to 77.77% than their respective miRNA homologs. Many of the target genes by the newly identified miRNAs were associated with plant growth and development, stress response, defense and hormone signaling, and oil synthesis pathways.</p><p><strong>Conclusion: </strong>These findings have the potential to speed up miRNA identification and expand our understanding of miRNA functions in J. curcas.</p>","PeriodicalId":74026,"journal":{"name":"Journal, genetic engineering & biotechnology","volume":"21 1","pages":"43"},"PeriodicalIF":2.8000,"publicationDate":"2023-04-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10079790/pdf/","citationCount":"0","resultStr":"{\"title\":\"Identification of conserved miRNAs and their targets in Jatropha curcas: an in silico approach.\",\"authors\":\"Foeaz Ahmed, Md Nazmul Islam Bappy, Md Shariful Islam\",\"doi\":\"10.1186/s43141-023-00495-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>MicroRNAs (miRNAs) are small endogenous RNAs with an approximate length of 18-22 nucleotides and involved in the regulation of gene expression in transcriptional or post-transcriptional levels. They were found to be associated with leaf morphogenesis, flowering time, vegetative phase change, and response to environmental cues in plants, where they act as a critical regulatory factor. The nature of high conservancy of plant miRNAs within the plant species made it possible to detect the conserved miRNAs by computational approaches. Expressed Sequence Tags (EST) based comparative genomic approaches provide advantages over wet lab approaches as it is convenient, easy to carry out and less time consuming. EST-based in silico approach can unravel new conserved miRNAs in plants, even when the complete genome sequence is not available.</p><p><strong>Results: </strong>To identify the novel miRNAs, a total of 46,865 ESTs from Jatropha curcas were searched for homology to all available 6746 mature miRNAs of plant eudicotyledons. Finally, we ended up with 12 novel miRNAs in Jatropha that range from 18 to 19 nucleotides where their respective precursor miRNAs had 54.11-71.76% (A + U) content. The putative miRNAs belong to 12 individual miRNA family and most of them have higher (A + U) content ranging from 47.36 to 77.77% than their respective miRNA homologs. Many of the target genes by the newly identified miRNAs were associated with plant growth and development, stress response, defense and hormone signaling, and oil synthesis pathways.</p><p><strong>Conclusion: </strong>These findings have the potential to speed up miRNA identification and expand our understanding of miRNA functions in J. curcas.</p>\",\"PeriodicalId\":74026,\"journal\":{\"name\":\"Journal, genetic engineering & biotechnology\",\"volume\":\"21 1\",\"pages\":\"43\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2023-04-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10079790/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal, genetic engineering & biotechnology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s43141-023-00495-9\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal, genetic engineering & biotechnology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s43141-023-00495-9","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
背景:MicroRNAs (miRNAs)是一种小的内源性rna,长度约为18-22个核苷酸,在转录或转录后水平上参与基因表达的调控。它们被发现与植物的叶片形态发生、开花时间、营养阶段变化和对环境信号的反应有关,在这些方面它们是一个关键的调节因子。植物mirna在植物物种中的高度保护性使得通过计算方法检测保守的mirna成为可能。基于表达序列标签(EST)的比较基因组方法提供了湿实验室方法的优势,因为它方便,易于实施,耗时更少。即使没有完整的基因组序列,基于est的芯片方法也可以揭示植物中新的保守mirna。结果:为了鉴定新的miRNAs,我们从麻疯树中检索了46,865条est序列,与植物真子叶中所有可用的6746条成熟miRNAs同源性。最后,我们在麻疯树中获得了12个新的mirna,范围从18到19个核苷酸,它们各自的前体mirna含量为54.11-71.76% (A + U)。推测的miRNA属于12个独立的miRNA家族,大多数miRNA的(A + U)含量高于各自的miRNA同源物,含量在47.36 ~ 77.77%之间。新发现的mirna的许多靶基因与植物生长发育、胁迫反应、防御和激素信号传导以及油脂合成途径有关。结论:这些发现有可能加速miRNA的鉴定,并扩大我们对麻瓜中miRNA功能的理解。
Identification of conserved miRNAs and their targets in Jatropha curcas: an in silico approach.
Background: MicroRNAs (miRNAs) are small endogenous RNAs with an approximate length of 18-22 nucleotides and involved in the regulation of gene expression in transcriptional or post-transcriptional levels. They were found to be associated with leaf morphogenesis, flowering time, vegetative phase change, and response to environmental cues in plants, where they act as a critical regulatory factor. The nature of high conservancy of plant miRNAs within the plant species made it possible to detect the conserved miRNAs by computational approaches. Expressed Sequence Tags (EST) based comparative genomic approaches provide advantages over wet lab approaches as it is convenient, easy to carry out and less time consuming. EST-based in silico approach can unravel new conserved miRNAs in plants, even when the complete genome sequence is not available.
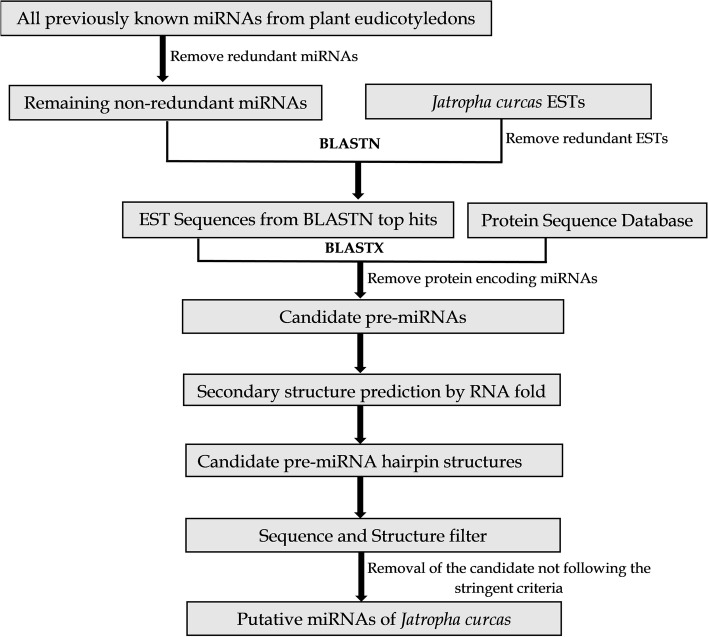
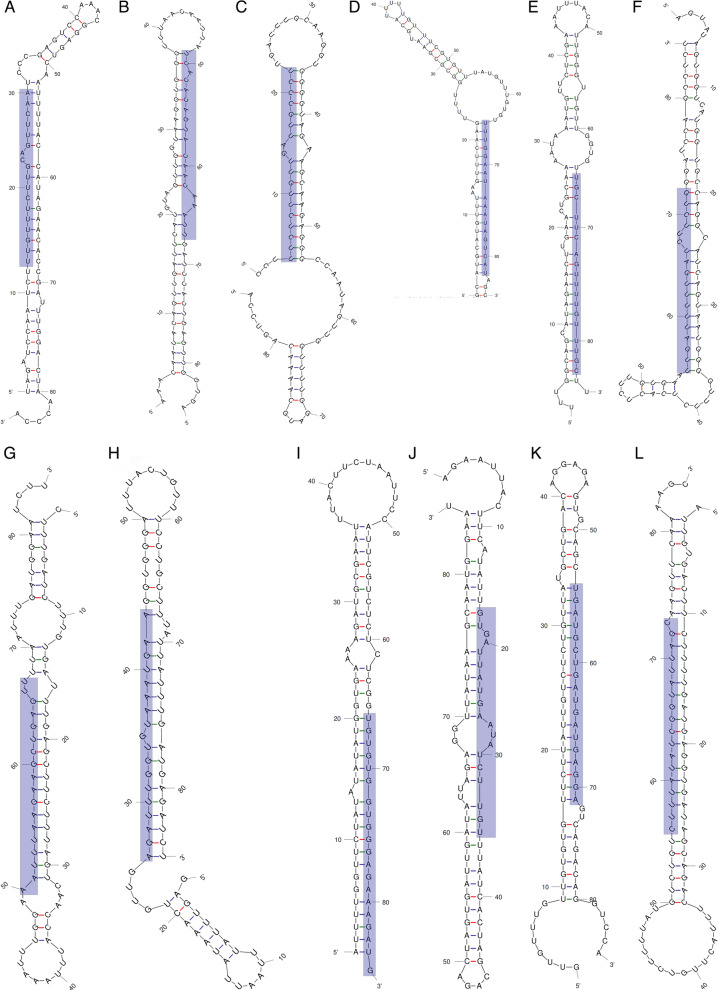
Results: To identify the novel miRNAs, a total of 46,865 ESTs from Jatropha curcas were searched for homology to all available 6746 mature miRNAs of plant eudicotyledons. Finally, we ended up with 12 novel miRNAs in Jatropha that range from 18 to 19 nucleotides where their respective precursor miRNAs had 54.11-71.76% (A + U) content. The putative miRNAs belong to 12 individual miRNA family and most of them have higher (A + U) content ranging from 47.36 to 77.77% than their respective miRNA homologs. Many of the target genes by the newly identified miRNAs were associated with plant growth and development, stress response, defense and hormone signaling, and oil synthesis pathways.
Conclusion: These findings have the potential to speed up miRNA identification and expand our understanding of miRNA functions in J. curcas.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


