Shruti Menon, Daniel Gracilla, Marcus R Breese, Yone Phar Lin, Filemon Dela Cruz, Tamar Feinberg, Elisa de Stanchina, Ana-Florina Galic, Hannah Allegakoen, Shruthi Perati, Nicholas Wen, Ann Heslin, Max A Horlbeck, Jonathan Weissman, E Alejandro Sweet-Cordero, Trever G Bivona, Asmin Tulpule
{"title":"FET融合癌蛋白破坏癌症的生理DNA修复网络。","authors":"Shruti Menon, Daniel Gracilla, Marcus R Breese, Yone Phar Lin, Filemon Dela Cruz, Tamar Feinberg, Elisa de Stanchina, Ana-Florina Galic, Hannah Allegakoen, Shruthi Perati, Nicholas Wen, Ann Heslin, Max A Horlbeck, Jonathan Weissman, E Alejandro Sweet-Cordero, Trever G Bivona, Asmin Tulpule","doi":"10.1101/2023.04.30.538578","DOIUrl":null,"url":null,"abstract":"<p><p>While oncogenes promote cancer cell growth, unrestrained proliferation represents a significant stressor to cellular homeostasis networks such as the DNA damage response (DDR). To enable oncogene tolerance, many cancers disable tumor suppressive DDR signaling through genetic loss of DDR pathways and downstream effectors (e.g., ATM or p53 tumor suppressor mutations). Whether and how oncogenes can help \"self-tolerize\" by creating analogous functional defects in physiologic DDR networks is not known. Here we focus on Ewing sarcoma, a FET fusion oncoprotein (EWSR1-FLI1) driven pediatric bone tumor, as a model for the class of FET rearranged cancers. Native FET family members are among the earliest factors recruited to DNA double-strand breaks (DSBs), though the function of both native FET proteins and FET fusion oncoproteins in DNA repair remains to be defined. We discover that the EWSR1-FLI1 fusion oncoprotein is recruited to DNA DSBs and interferes with native FET (EWSR1) protein function in activating the DNA damage sensor ATM. In multiple FET rearranged cancers, FET fusion oncoproteins induce functional ATM defects, rendering the compensatory ATR signaling axis as a collateral dependency and therapeutic target. More generally, we find that aberrant recruitment of a fusion oncoprotein to sites of DNA damage can disrupt physiologic DSB repair, revealing a mechanism for how growth-promoting oncogenes can also create functional defects within tumor suppressive DDR networks.</p>","PeriodicalId":72407,"journal":{"name":"bioRxiv : the preprint server for biology","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-11-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1e/db/nihpp-2023.04.30.538578v2.PMC10187251.pdf","citationCount":"0","resultStr":"{\"title\":\"FET fusion oncoproteins disrupt physiologic DNA repair networks in cancer.\",\"authors\":\"Shruti Menon, Daniel Gracilla, Marcus R Breese, Yone Phar Lin, Filemon Dela Cruz, Tamar Feinberg, Elisa de Stanchina, Ana-Florina Galic, Hannah Allegakoen, Shruthi Perati, Nicholas Wen, Ann Heslin, Max A Horlbeck, Jonathan Weissman, E Alejandro Sweet-Cordero, Trever G Bivona, Asmin Tulpule\",\"doi\":\"10.1101/2023.04.30.538578\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>While oncogenes promote cancer cell growth, unrestrained proliferation represents a significant stressor to cellular homeostasis networks such as the DNA damage response (DDR). To enable oncogene tolerance, many cancers disable tumor suppressive DDR signaling through genetic loss of DDR pathways and downstream effectors (e.g., ATM or p53 tumor suppressor mutations). Whether and how oncogenes can help \\\"self-tolerize\\\" by creating analogous functional defects in physiologic DDR networks is not known. Here we focus on Ewing sarcoma, a FET fusion oncoprotein (EWSR1-FLI1) driven pediatric bone tumor, as a model for the class of FET rearranged cancers. Native FET family members are among the earliest factors recruited to DNA double-strand breaks (DSBs), though the function of both native FET proteins and FET fusion oncoproteins in DNA repair remains to be defined. We discover that the EWSR1-FLI1 fusion oncoprotein is recruited to DNA DSBs and interferes with native FET (EWSR1) protein function in activating the DNA damage sensor ATM. In multiple FET rearranged cancers, FET fusion oncoproteins induce functional ATM defects, rendering the compensatory ATR signaling axis as a collateral dependency and therapeutic target. More generally, we find that aberrant recruitment of a fusion oncoprotein to sites of DNA damage can disrupt physiologic DSB repair, revealing a mechanism for how growth-promoting oncogenes can also create functional defects within tumor suppressive DDR networks.</p>\",\"PeriodicalId\":72407,\"journal\":{\"name\":\"bioRxiv : the preprint server for biology\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-11-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1e/db/nihpp-2023.04.30.538578v2.PMC10187251.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"bioRxiv : the preprint server for biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/2023.04.30.538578\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"bioRxiv : the preprint server for biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2023.04.30.538578","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
FET fusion oncoproteins disrupt physiologic DNA repair networks in cancer.
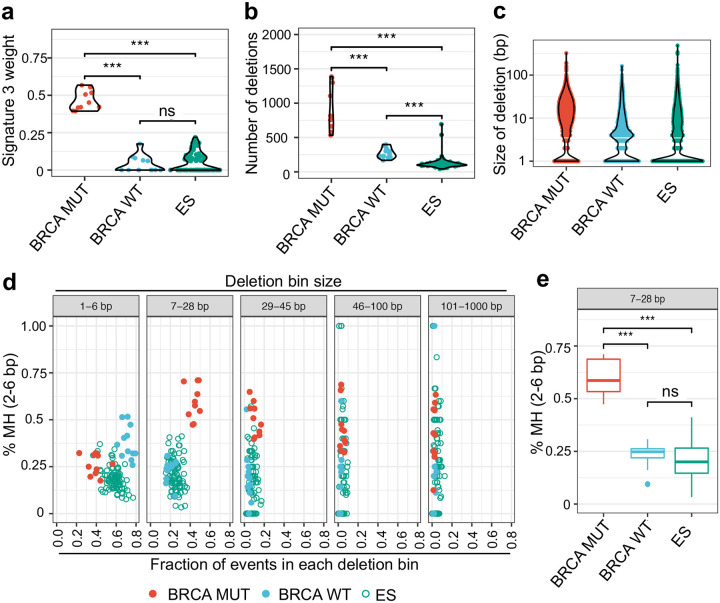
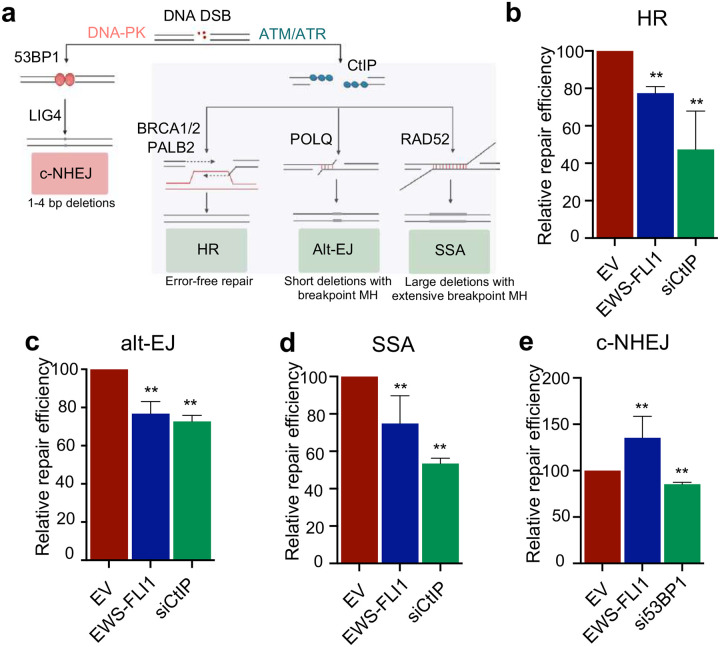
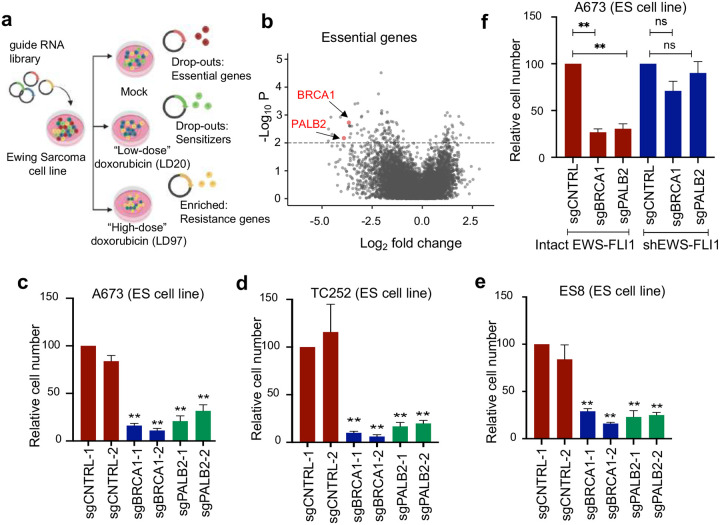
While oncogenes promote cancer cell growth, unrestrained proliferation represents a significant stressor to cellular homeostasis networks such as the DNA damage response (DDR). To enable oncogene tolerance, many cancers disable tumor suppressive DDR signaling through genetic loss of DDR pathways and downstream effectors (e.g., ATM or p53 tumor suppressor mutations). Whether and how oncogenes can help "self-tolerize" by creating analogous functional defects in physiologic DDR networks is not known. Here we focus on Ewing sarcoma, a FET fusion oncoprotein (EWSR1-FLI1) driven pediatric bone tumor, as a model for the class of FET rearranged cancers. Native FET family members are among the earliest factors recruited to DNA double-strand breaks (DSBs), though the function of both native FET proteins and FET fusion oncoproteins in DNA repair remains to be defined. We discover that the EWSR1-FLI1 fusion oncoprotein is recruited to DNA DSBs and interferes with native FET (EWSR1) protein function in activating the DNA damage sensor ATM. In multiple FET rearranged cancers, FET fusion oncoproteins induce functional ATM defects, rendering the compensatory ATR signaling axis as a collateral dependency and therapeutic target. More generally, we find that aberrant recruitment of a fusion oncoprotein to sites of DNA damage can disrupt physiologic DSB repair, revealing a mechanism for how growth-promoting oncogenes can also create functional defects within tumor suppressive DDR networks.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


