Anastasia Gromova, Byeonggu Cha, Erica M Robinson, Laura M Strickland, Nhat Nguyen, Mai K ElMallah, Constanza J Cortes, Albert R La Spada
{"title":"X 连锁 SBMA 模型小鼠表现出相关的非神经系统表型,其运动神经元中突变雄激素受体蛋白的表达并非神经肌肉疾病的必要条件。","authors":"Anastasia Gromova, Byeonggu Cha, Erica M Robinson, Laura M Strickland, Nhat Nguyen, Mai K ElMallah, Constanza J Cortes, Albert R La Spada","doi":"10.1186/s40478-023-01582-1","DOIUrl":null,"url":null,"abstract":"<p><p>X-linked spinal and bulbar muscular atrophy (SBMA; Kennedy's disease) is a rare neuromuscular disorder characterized by adult-onset proximal muscle weakness and lower motor neuron degeneration. SBMA was the first human disease found to be caused by a repeat expansion mutation, as affected patients possess an expanded tract of CAG repeats, encoding polyglutamine, in the androgen receptor (AR) gene. We previously developed a conditional BAC fxAR121 transgenic mouse model of SBMA and used it to define a primary role for skeletal muscle expression of polyglutamine-expanded AR in causing the motor neuron degeneration. Here we sought to extend our understanding of SBMA disease pathophysiology and cellular basis by detailed examination and directed experimentation with the BAC fxAR121 mice. First, we evaluated BAC fxAR121 mice for non-neurological disease phenotypes recently described in human SBMA patients, and documented prominent non-alcoholic fatty liver disease, cardiomegaly, and ventricular heart wall thinning in aged male BAC fxAR121 mice. Our discovery of significant hepatic and cardiac abnormalities in SBMA mice underscores the need to evaluate human SBMA patients for signs of liver and heart disease. To directly examine the contribution of motor neuron-expressed polyQ-AR protein to SBMA neurodegeneration, we crossed BAC fxAR121 mice with two different lines of transgenic mice expressing Cre recombinase in motor neurons, and after updating characterization of SBMA phenotypes in our current BAC fxAR121 colony, we found that excision of mutant AR from motor neurons did not rescue neuromuscular or systemic disease. These findings further validate a primary role for skeletal muscle as the driver of SBMA motor neuronopathy and indicate that therapies being developed to treat patients should be delivered peripherally.</p>","PeriodicalId":6914,"journal":{"name":"Acta Neuropathologica Communications","volume":"11 1","pages":"90"},"PeriodicalIF":6.2000,"publicationDate":"2023-06-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10239133/pdf/","citationCount":"0","resultStr":"{\"title\":\"X-linked SBMA model mice display relevant non-neurological phenotypes and their expression of mutant androgen receptor protein in motor neurons is not required for neuromuscular disease.\",\"authors\":\"Anastasia Gromova, Byeonggu Cha, Erica M Robinson, Laura M Strickland, Nhat Nguyen, Mai K ElMallah, Constanza J Cortes, Albert R La Spada\",\"doi\":\"10.1186/s40478-023-01582-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>X-linked spinal and bulbar muscular atrophy (SBMA; Kennedy's disease) is a rare neuromuscular disorder characterized by adult-onset proximal muscle weakness and lower motor neuron degeneration. SBMA was the first human disease found to be caused by a repeat expansion mutation, as affected patients possess an expanded tract of CAG repeats, encoding polyglutamine, in the androgen receptor (AR) gene. We previously developed a conditional BAC fxAR121 transgenic mouse model of SBMA and used it to define a primary role for skeletal muscle expression of polyglutamine-expanded AR in causing the motor neuron degeneration. Here we sought to extend our understanding of SBMA disease pathophysiology and cellular basis by detailed examination and directed experimentation with the BAC fxAR121 mice. First, we evaluated BAC fxAR121 mice for non-neurological disease phenotypes recently described in human SBMA patients, and documented prominent non-alcoholic fatty liver disease, cardiomegaly, and ventricular heart wall thinning in aged male BAC fxAR121 mice. Our discovery of significant hepatic and cardiac abnormalities in SBMA mice underscores the need to evaluate human SBMA patients for signs of liver and heart disease. To directly examine the contribution of motor neuron-expressed polyQ-AR protein to SBMA neurodegeneration, we crossed BAC fxAR121 mice with two different lines of transgenic mice expressing Cre recombinase in motor neurons, and after updating characterization of SBMA phenotypes in our current BAC fxAR121 colony, we found that excision of mutant AR from motor neurons did not rescue neuromuscular or systemic disease. These findings further validate a primary role for skeletal muscle as the driver of SBMA motor neuronopathy and indicate that therapies being developed to treat patients should be delivered peripherally.</p>\",\"PeriodicalId\":6914,\"journal\":{\"name\":\"Acta Neuropathologica Communications\",\"volume\":\"11 1\",\"pages\":\"90\"},\"PeriodicalIF\":6.2000,\"publicationDate\":\"2023-06-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10239133/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Neuropathologica Communications\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s40478-023-01582-1\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"NEUROSCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Neuropathologica Communications","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40478-023-01582-1","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
X-linked SBMA model mice display relevant non-neurological phenotypes and their expression of mutant androgen receptor protein in motor neurons is not required for neuromuscular disease.
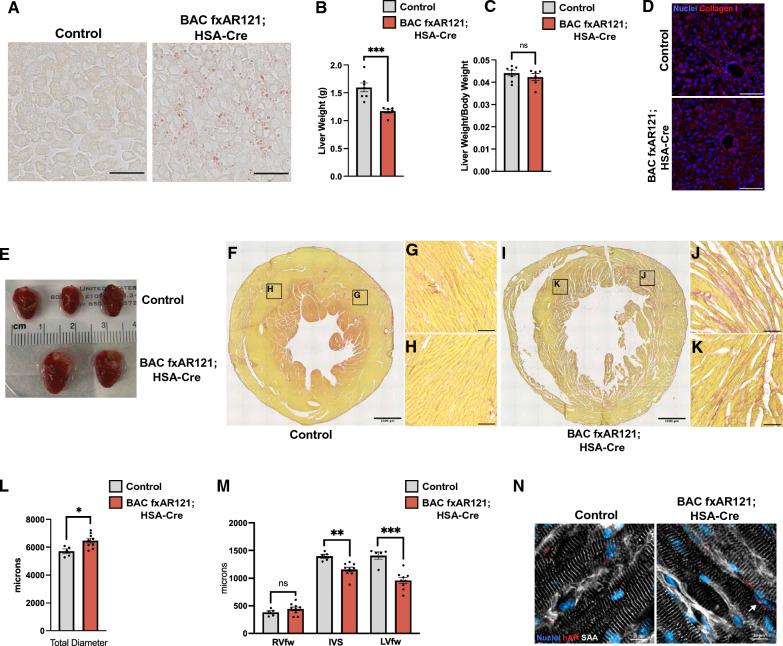
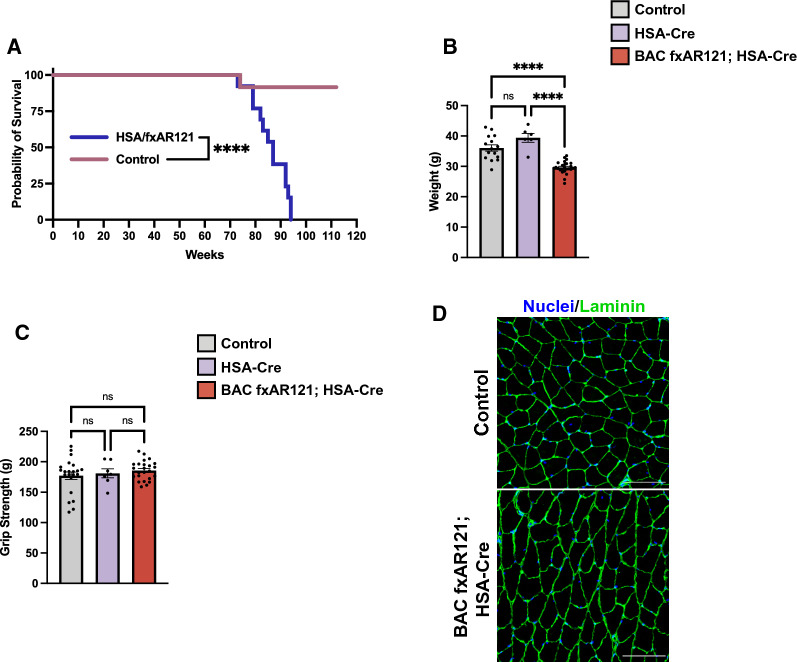
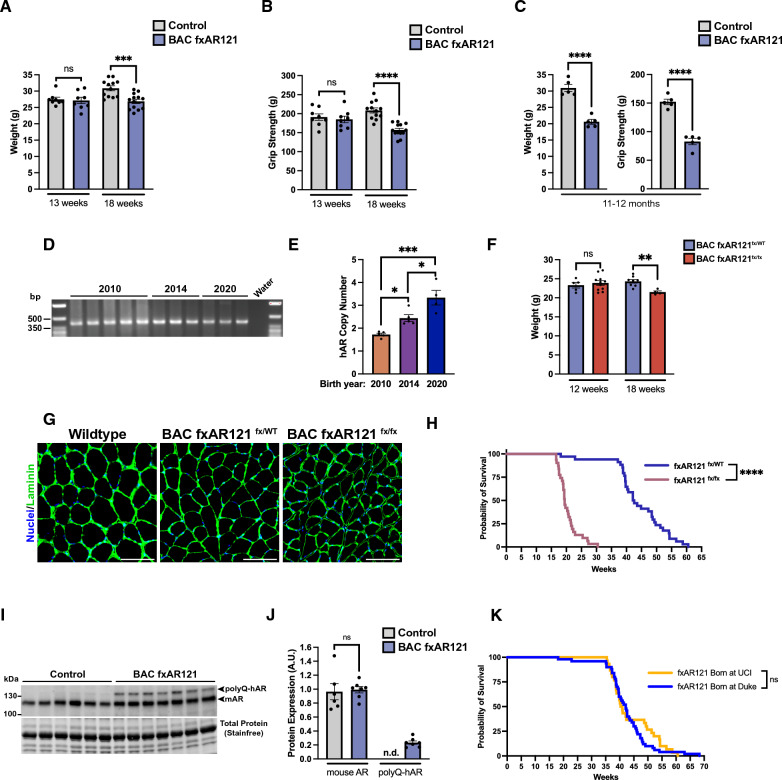
X-linked spinal and bulbar muscular atrophy (SBMA; Kennedy's disease) is a rare neuromuscular disorder characterized by adult-onset proximal muscle weakness and lower motor neuron degeneration. SBMA was the first human disease found to be caused by a repeat expansion mutation, as affected patients possess an expanded tract of CAG repeats, encoding polyglutamine, in the androgen receptor (AR) gene. We previously developed a conditional BAC fxAR121 transgenic mouse model of SBMA and used it to define a primary role for skeletal muscle expression of polyglutamine-expanded AR in causing the motor neuron degeneration. Here we sought to extend our understanding of SBMA disease pathophysiology and cellular basis by detailed examination and directed experimentation with the BAC fxAR121 mice. First, we evaluated BAC fxAR121 mice for non-neurological disease phenotypes recently described in human SBMA patients, and documented prominent non-alcoholic fatty liver disease, cardiomegaly, and ventricular heart wall thinning in aged male BAC fxAR121 mice. Our discovery of significant hepatic and cardiac abnormalities in SBMA mice underscores the need to evaluate human SBMA patients for signs of liver and heart disease. To directly examine the contribution of motor neuron-expressed polyQ-AR protein to SBMA neurodegeneration, we crossed BAC fxAR121 mice with two different lines of transgenic mice expressing Cre recombinase in motor neurons, and after updating characterization of SBMA phenotypes in our current BAC fxAR121 colony, we found that excision of mutant AR from motor neurons did not rescue neuromuscular or systemic disease. These findings further validate a primary role for skeletal muscle as the driver of SBMA motor neuronopathy and indicate that therapies being developed to treat patients should be delivered peripherally.
期刊介绍:
"Acta Neuropathologica Communications (ANC)" is a peer-reviewed journal that specializes in the rapid publication of research articles focused on the mechanisms underlying neurological diseases. The journal emphasizes the use of molecular, cellular, and morphological techniques applied to experimental or human tissues to investigate the pathogenesis of neurological disorders.
ANC is committed to a fast-track publication process, aiming to publish accepted manuscripts within two months of submission. This expedited timeline is designed to ensure that the latest findings in neuroscience and pathology are disseminated quickly to the scientific community, fostering rapid advancements in the field of neurology and neuroscience. The journal's focus on cutting-edge research and its swift publication schedule make it a valuable resource for researchers, clinicians, and other professionals interested in the study and treatment of neurological conditions.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


