David Stevens, Shadi Milani-Nejad, Tahseen Mozaffar
{"title":"庞贝病:临床、诊断和治疗综述。","authors":"David Stevens, Shadi Milani-Nejad, Tahseen Mozaffar","doi":"10.1007/s11940-022-00736-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose of review: </strong>This review summarizes the clinical presentation and provides an update on the current strategies for diagnosis of Pompe disease. We will review the available treatment options. We examine newly approved treatments as well as upcoming therapies in this condition. We also provide commentary on the unmet needs in clinical management and research for this disease.</p><p><strong>Recent findings: </strong>In March 2015, Pompe disease was added to the Recommended Uniform Screening Panel (RUSP) and since then a number of states have added Pompe disease to their slate of diseases for their Newborn Screening (NBS) program. Data emerging from these programs is revising our knowledge of incidence of Pompe disease. In 2021, two randomized controlled trials involving new forms of enzyme replacement therapy (ERT) were completed and one new product is already FDA-approved and on the market, whereas the other product will come up for FDA review in the fall. Neither of the new ERT were shown to be superior to the standard of care product, <i>alglucosidase</i>. The long-term effectiveness of these newer forms of ERT is unclear. Newer versions of the ERT are in development in addition to multiple different strategies of gene therapy to deliver GAA, the gene responsible for producing acid alpha-glucosidase, the defective protein in Pompe Disease. Glycogen substrate reduction is also in development in Pompe disease and other glycogen storage disorders.</p><p><strong>Summary: </strong>There are significant unmet needs as it relates to clinical care and therapeutics in Pompe disease as well as in research. The currently available treatments lose effectiveness over the long run and do not have penetration into neuronal tissues and inconsistent penetration in certain muscles. More definitive gene therapy and enzyme replacement strategies are currently in development and testing.</p>","PeriodicalId":10975,"journal":{"name":"Current Treatment Options in Neurology","volume":"24 11","pages":"573-588"},"PeriodicalIF":1.8000,"publicationDate":"2022-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10035871/pdf/","citationCount":"7","resultStr":"{\"title\":\"Pompe Disease: a Clinical, Diagnostic, and Therapeutic Overview.\",\"authors\":\"David Stevens, Shadi Milani-Nejad, Tahseen Mozaffar\",\"doi\":\"10.1007/s11940-022-00736-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Purpose of review: </strong>This review summarizes the clinical presentation and provides an update on the current strategies for diagnosis of Pompe disease. We will review the available treatment options. We examine newly approved treatments as well as upcoming therapies in this condition. We also provide commentary on the unmet needs in clinical management and research for this disease.</p><p><strong>Recent findings: </strong>In March 2015, Pompe disease was added to the Recommended Uniform Screening Panel (RUSP) and since then a number of states have added Pompe disease to their slate of diseases for their Newborn Screening (NBS) program. Data emerging from these programs is revising our knowledge of incidence of Pompe disease. In 2021, two randomized controlled trials involving new forms of enzyme replacement therapy (ERT) were completed and one new product is already FDA-approved and on the market, whereas the other product will come up for FDA review in the fall. Neither of the new ERT were shown to be superior to the standard of care product, <i>alglucosidase</i>. The long-term effectiveness of these newer forms of ERT is unclear. Newer versions of the ERT are in development in addition to multiple different strategies of gene therapy to deliver GAA, the gene responsible for producing acid alpha-glucosidase, the defective protein in Pompe Disease. Glycogen substrate reduction is also in development in Pompe disease and other glycogen storage disorders.</p><p><strong>Summary: </strong>There are significant unmet needs as it relates to clinical care and therapeutics in Pompe disease as well as in research. The currently available treatments lose effectiveness over the long run and do not have penetration into neuronal tissues and inconsistent penetration in certain muscles. More definitive gene therapy and enzyme replacement strategies are currently in development and testing.</p>\",\"PeriodicalId\":10975,\"journal\":{\"name\":\"Current Treatment Options in Neurology\",\"volume\":\"24 11\",\"pages\":\"573-588\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2022-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10035871/pdf/\",\"citationCount\":\"7\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current Treatment Options in Neurology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s11940-022-00736-1\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Treatment Options in Neurology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s11940-022-00736-1","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
Pompe Disease: a Clinical, Diagnostic, and Therapeutic Overview.
Purpose of review: This review summarizes the clinical presentation and provides an update on the current strategies for diagnosis of Pompe disease. We will review the available treatment options. We examine newly approved treatments as well as upcoming therapies in this condition. We also provide commentary on the unmet needs in clinical management and research for this disease.
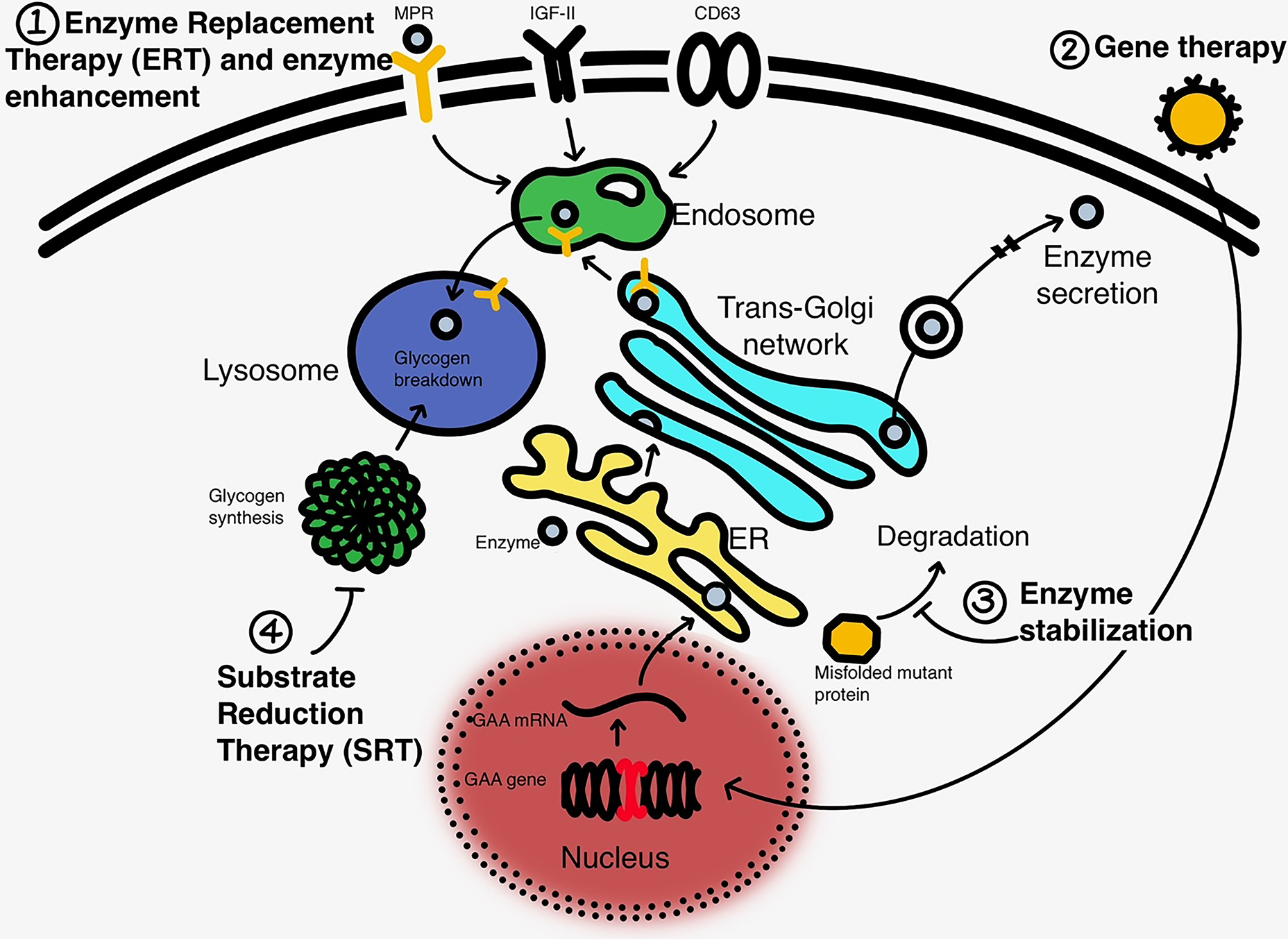
Recent findings: In March 2015, Pompe disease was added to the Recommended Uniform Screening Panel (RUSP) and since then a number of states have added Pompe disease to their slate of diseases for their Newborn Screening (NBS) program. Data emerging from these programs is revising our knowledge of incidence of Pompe disease. In 2021, two randomized controlled trials involving new forms of enzyme replacement therapy (ERT) were completed and one new product is already FDA-approved and on the market, whereas the other product will come up for FDA review in the fall. Neither of the new ERT were shown to be superior to the standard of care product, alglucosidase. The long-term effectiveness of these newer forms of ERT is unclear. Newer versions of the ERT are in development in addition to multiple different strategies of gene therapy to deliver GAA, the gene responsible for producing acid alpha-glucosidase, the defective protein in Pompe Disease. Glycogen substrate reduction is also in development in Pompe disease and other glycogen storage disorders.
Summary: There are significant unmet needs as it relates to clinical care and therapeutics in Pompe disease as well as in research. The currently available treatments lose effectiveness over the long run and do not have penetration into neuronal tissues and inconsistent penetration in certain muscles. More definitive gene therapy and enzyme replacement strategies are currently in development and testing.
期刊介绍:
This journal aims to review the most important, recently published treatment option advances in the field of neurology. By presenting clear, insightful, balanced contributions by international experts, the journal intends to facilitate worldwide approaches to the treatment of neurologic conditions.
We accomplish this aim by appointing international authorities to serve as Section Editors in key subject areas, such as epilepsy, headache, neurologic ophthalmology and otology, neuromuscular disorders, psychiatric manifestations of neurologic disease, and sleep disorders. Section Editors select topics for which leading experts contribute comprehensive review articles that emphasize new developments and recently published papers of major importance, highlighted by annotated reference lists. We also provide commentaries from well-known neurologists, and an international Editorial Board reviews the annual table of contents, suggests articles of special interest to their country/region, and ensures that topics are current and include emerging research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


