Gregory J. M. Zajac, Sarah A. Gagliano Taliun, Carlo Sidore, Sarah E. Graham, Bjørn O. Åsvold, Ben Brumpton, Jonas B. Nielsen, Wei Zhou, Maiken Gabrielsen, Anne H. Skogholt, Lars G. Fritsche, David Schlessinger, Francesco Cucca, Kristian Hveem, Cristen J. Willer, Gonçalo R. Abecasis
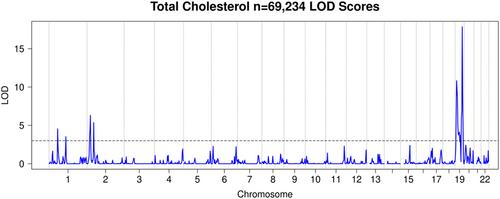
{"title":"GWAS群体相关个体的快速连锁分析方法","authors":"Gregory J. M. Zajac, Sarah A. Gagliano Taliun, Carlo Sidore, Sarah E. Graham, Bjørn O. Åsvold, Ben Brumpton, Jonas B. Nielsen, Wei Zhou, Maiken Gabrielsen, Anne H. Skogholt, Lars G. Fritsche, David Schlessinger, Francesco Cucca, Kristian Hveem, Cristen J. Willer, Gonçalo R. Abecasis","doi":"10.1002/gepi.22516","DOIUrl":null,"url":null,"abstract":"<p>Linkage analysis, a class of methods for detecting co-segregation of genomic segments and traits in families, was used to map disease-causing genes for decades before genotyping arrays and dense SNP genotyping enabled genome-wide association studies in population samples. Population samples often contain related individuals, but the segregation of alleles within families is rarely used because traditional linkage methods are computationally inefficient for larger datasets. Here, we describe Population Linkage, a novel application of Haseman–Elston regression as a method of moments estimator of variance components and their standard errors. We achieve additional computational efficiency by using modern methods for detection of IBD segments and variance component estimation, efficient preprocessing of input data, and minimizing redundant numerical calculations. We also refined variance component models to account for the biases in population-scale methods for IBD segment detection. We ran Population Linkage on four blood lipid traits in over 70,000 individuals from the HUNT and SardiNIA studies, successfully detecting 25 known genetic signals. One notable linkage signal that appeared in both was for low-density lipoprotein (LDL) cholesterol levels in the region near the gene <i>APOE</i> (LOD = 29.3, variance explained = 4.1%). This is the region where the missense variants rs7412 and rs429358, which together make up the ε2, ε3, and ε4 alleles each account for 2.4% and 0.8% of variation in circulating LDL cholesterol. Our results show the potential for linkage analysis and other large-scale applications of method of moments variance components estimation.</p>","PeriodicalId":12710,"journal":{"name":"Genetic Epidemiology","volume":"47 3","pages":"231-248"},"PeriodicalIF":3.8000,"publicationDate":"2023-02-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/gepi.22516","citationCount":"0","resultStr":"{\"title\":\"A fast linkage method for population GWAS cohorts with related individuals\",\"authors\":\"Gregory J. M. Zajac, Sarah A. Gagliano Taliun, Carlo Sidore, Sarah E. Graham, Bjørn O. Åsvold, Ben Brumpton, Jonas B. Nielsen, Wei Zhou, Maiken Gabrielsen, Anne H. Skogholt, Lars G. Fritsche, David Schlessinger, Francesco Cucca, Kristian Hveem, Cristen J. Willer, Gonçalo R. Abecasis\",\"doi\":\"10.1002/gepi.22516\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Linkage analysis, a class of methods for detecting co-segregation of genomic segments and traits in families, was used to map disease-causing genes for decades before genotyping arrays and dense SNP genotyping enabled genome-wide association studies in population samples. Population samples often contain related individuals, but the segregation of alleles within families is rarely used because traditional linkage methods are computationally inefficient for larger datasets. Here, we describe Population Linkage, a novel application of Haseman–Elston regression as a method of moments estimator of variance components and their standard errors. We achieve additional computational efficiency by using modern methods for detection of IBD segments and variance component estimation, efficient preprocessing of input data, and minimizing redundant numerical calculations. We also refined variance component models to account for the biases in population-scale methods for IBD segment detection. We ran Population Linkage on four blood lipid traits in over 70,000 individuals from the HUNT and SardiNIA studies, successfully detecting 25 known genetic signals. One notable linkage signal that appeared in both was for low-density lipoprotein (LDL) cholesterol levels in the region near the gene <i>APOE</i> (LOD = 29.3, variance explained = 4.1%). This is the region where the missense variants rs7412 and rs429358, which together make up the ε2, ε3, and ε4 alleles each account for 2.4% and 0.8% of variation in circulating LDL cholesterol. Our results show the potential for linkage analysis and other large-scale applications of method of moments variance components estimation.</p>\",\"PeriodicalId\":12710,\"journal\":{\"name\":\"Genetic Epidemiology\",\"volume\":\"47 3\",\"pages\":\"231-248\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2023-02-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/gepi.22516\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genetic Epidemiology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/gepi.22516\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetic Epidemiology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/gepi.22516","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
A fast linkage method for population GWAS cohorts with related individuals
Linkage analysis, a class of methods for detecting co-segregation of genomic segments and traits in families, was used to map disease-causing genes for decades before genotyping arrays and dense SNP genotyping enabled genome-wide association studies in population samples. Population samples often contain related individuals, but the segregation of alleles within families is rarely used because traditional linkage methods are computationally inefficient for larger datasets. Here, we describe Population Linkage, a novel application of Haseman–Elston regression as a method of moments estimator of variance components and their standard errors. We achieve additional computational efficiency by using modern methods for detection of IBD segments and variance component estimation, efficient preprocessing of input data, and minimizing redundant numerical calculations. We also refined variance component models to account for the biases in population-scale methods for IBD segment detection. We ran Population Linkage on four blood lipid traits in over 70,000 individuals from the HUNT and SardiNIA studies, successfully detecting 25 known genetic signals. One notable linkage signal that appeared in both was for low-density lipoprotein (LDL) cholesterol levels in the region near the gene APOE (LOD = 29.3, variance explained = 4.1%). This is the region where the missense variants rs7412 and rs429358, which together make up the ε2, ε3, and ε4 alleles each account for 2.4% and 0.8% of variation in circulating LDL cholesterol. Our results show the potential for linkage analysis and other large-scale applications of method of moments variance components estimation.
期刊介绍:
Genetic Epidemiology is a peer-reviewed journal for discussion of research on the genetic causes of the distribution of human traits in families and populations. Emphasis is placed on the relative contribution of genetic and environmental factors to human disease as revealed by genetic, epidemiological, and biologic investigations.
Genetic Epidemiology primarily publishes papers in statistical genetics, a research field that is primarily concerned with development of statistical, bioinformatical, and computational models for analyzing genetic data. Incorporation of underlying biology and population genetics into conceptual models is favored. The Journal seeks original articles comprising either applied research or innovative statistical, mathematical, computational, or genomic methodologies that advance studies in genetic epidemiology. Other types of reports are encouraged, such as letters to the editor, topic reviews, and perspectives from other fields of research that will likely enrich the field of genetic epidemiology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


