{"title":"利用分层化学图表示和多分辨率图变分自编码器的基于片段的深度分子生成。","authors":"Zhenxiang Gao, Xinyu Wang, Blake Blumenfeld Gaines, Xuetao Shi, Jinbo Bi, Minghu Song","doi":"10.1002/minf.202200215","DOIUrl":null,"url":null,"abstract":"<p><p>Graph generative models have recently emerged as an interesting approach to construct molecular structures atom-by-atom or fragment-by-fragment. In this study, we adopt the fragment-based strategy and decompose each input molecule into a set of small chemical fragments. In drug discovery, a few drug molecules are designed by replacing certain chemical substituents with their bioisosteres or alternative chemical moieties. This inspires us to group decomposed fragments into different fragment clusters according to their local structural environment around bond-breaking positions. In this way, an input structure can be transformed into an equivalent three-layer graph, in which individual atoms, decomposed fragments, or obtained fragment clusters act as graph nodes at each corresponding layer. We further implement a prototype model, named multi-resolution graph variational autoencoder (MRGVAE), to learn embeddings of constituted nodes at each layer in a fine-to-coarse order. Our decoder adopts a similar but conversely hierarchical structure. It first predicts the next possible fragment cluster, then samples an exact fragment structure out of the determined fragment cluster, and sequentially attaches it to the preceding chemical moiety. Our proposed approach demonstrates comparatively good performance in molecular evaluation metrics compared with several other graph-based molecular generative models. The introduction of the additional fragment cluster graph layer will hopefully increase the odds of assembling new chemical moieties absent in the original training set and enhance their structural diversity. We hope that our prototyping work will inspire more creative research to explore the possibility of incorporating different kinds of chemical domain knowledge into a similar multi-resolution neural network architecture.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":"42 5","pages":"e2200215"},"PeriodicalIF":2.8000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Fragment-based deep molecular generation using hierarchical chemical graph representation and multi-resolution graph variational autoencoder.\",\"authors\":\"Zhenxiang Gao, Xinyu Wang, Blake Blumenfeld Gaines, Xuetao Shi, Jinbo Bi, Minghu Song\",\"doi\":\"10.1002/minf.202200215\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Graph generative models have recently emerged as an interesting approach to construct molecular structures atom-by-atom or fragment-by-fragment. In this study, we adopt the fragment-based strategy and decompose each input molecule into a set of small chemical fragments. In drug discovery, a few drug molecules are designed by replacing certain chemical substituents with their bioisosteres or alternative chemical moieties. This inspires us to group decomposed fragments into different fragment clusters according to their local structural environment around bond-breaking positions. In this way, an input structure can be transformed into an equivalent three-layer graph, in which individual atoms, decomposed fragments, or obtained fragment clusters act as graph nodes at each corresponding layer. We further implement a prototype model, named multi-resolution graph variational autoencoder (MRGVAE), to learn embeddings of constituted nodes at each layer in a fine-to-coarse order. Our decoder adopts a similar but conversely hierarchical structure. It first predicts the next possible fragment cluster, then samples an exact fragment structure out of the determined fragment cluster, and sequentially attaches it to the preceding chemical moiety. Our proposed approach demonstrates comparatively good performance in molecular evaluation metrics compared with several other graph-based molecular generative models. The introduction of the additional fragment cluster graph layer will hopefully increase the odds of assembling new chemical moieties absent in the original training set and enhance their structural diversity. We hope that our prototyping work will inspire more creative research to explore the possibility of incorporating different kinds of chemical domain knowledge into a similar multi-resolution neural network architecture.</p>\",\"PeriodicalId\":18853,\"journal\":{\"name\":\"Molecular Informatics\",\"volume\":\"42 5\",\"pages\":\"e2200215\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2023-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Informatics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/minf.202200215\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202200215","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Fragment-based deep molecular generation using hierarchical chemical graph representation and multi-resolution graph variational autoencoder.
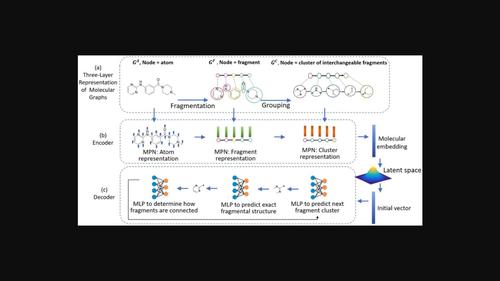
Graph generative models have recently emerged as an interesting approach to construct molecular structures atom-by-atom or fragment-by-fragment. In this study, we adopt the fragment-based strategy and decompose each input molecule into a set of small chemical fragments. In drug discovery, a few drug molecules are designed by replacing certain chemical substituents with their bioisosteres or alternative chemical moieties. This inspires us to group decomposed fragments into different fragment clusters according to their local structural environment around bond-breaking positions. In this way, an input structure can be transformed into an equivalent three-layer graph, in which individual atoms, decomposed fragments, or obtained fragment clusters act as graph nodes at each corresponding layer. We further implement a prototype model, named multi-resolution graph variational autoencoder (MRGVAE), to learn embeddings of constituted nodes at each layer in a fine-to-coarse order. Our decoder adopts a similar but conversely hierarchical structure. It first predicts the next possible fragment cluster, then samples an exact fragment structure out of the determined fragment cluster, and sequentially attaches it to the preceding chemical moiety. Our proposed approach demonstrates comparatively good performance in molecular evaluation metrics compared with several other graph-based molecular generative models. The introduction of the additional fragment cluster graph layer will hopefully increase the odds of assembling new chemical moieties absent in the original training set and enhance their structural diversity. We hope that our prototyping work will inspire more creative research to explore the possibility of incorporating different kinds of chemical domain knowledge into a similar multi-resolution neural network architecture.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


