{"title":"使用 AlphaFold2 和 DAQ 分数完善低温电子显微镜图的蛋白质模型。","authors":"Genki Terashi, Xiao Wang, Daisuke Kihara","doi":"10.1107/S2059798322011676","DOIUrl":null,"url":null,"abstract":"<p><p>As more protein structure models have been determined from cryogenic electron microscopy (cryo-EM) density maps, establishing how to evaluate the model accuracy and how to correct models in cases where they contain errors is becoming crucial to ensure the quality of the structural models deposited in the public database, the PDB. Here, a new protocol is presented for evaluating a protein model built from a cryo-EM map and applying local structure refinement in the case where the model has potential errors. Firstly, model evaluation is performed using a deep-learning-based model-local map assessment score, DAQ, that has recently been developed. The subsequent local refinement is performed by a modified AlphaFold2 procedure, in which a trimmed template model and a trimmed multiple sequence alignment are provided as input to control which structure regions to refine while leaving other more confident regions of the model intact. A benchmark study showed that this protocol, DAQ-refine, consistently improves low-quality regions of the initial models. Among 18 refined models generated for an initial structure, DAQ shows a high correlation with model quality and can identify the best accurate model for most of the tested cases. The improvements obtained by DAQ-refine were on average larger than other existing methods.</p>","PeriodicalId":7116,"journal":{"name":"Acta Crystallographica. Section D, Structural Biology","volume":"79 Pt 1","pages":"10-21"},"PeriodicalIF":2.6000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9815095/pdf/","citationCount":"0","resultStr":"{\"title\":\"Protein model refinement for cryo-EM maps using AlphaFold2 and the DAQ score.\",\"authors\":\"Genki Terashi, Xiao Wang, Daisuke Kihara\",\"doi\":\"10.1107/S2059798322011676\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>As more protein structure models have been determined from cryogenic electron microscopy (cryo-EM) density maps, establishing how to evaluate the model accuracy and how to correct models in cases where they contain errors is becoming crucial to ensure the quality of the structural models deposited in the public database, the PDB. Here, a new protocol is presented for evaluating a protein model built from a cryo-EM map and applying local structure refinement in the case where the model has potential errors. Firstly, model evaluation is performed using a deep-learning-based model-local map assessment score, DAQ, that has recently been developed. The subsequent local refinement is performed by a modified AlphaFold2 procedure, in which a trimmed template model and a trimmed multiple sequence alignment are provided as input to control which structure regions to refine while leaving other more confident regions of the model intact. A benchmark study showed that this protocol, DAQ-refine, consistently improves low-quality regions of the initial models. Among 18 refined models generated for an initial structure, DAQ shows a high correlation with model quality and can identify the best accurate model for most of the tested cases. The improvements obtained by DAQ-refine were on average larger than other existing methods.</p>\",\"PeriodicalId\":7116,\"journal\":{\"name\":\"Acta Crystallographica. Section D, Structural Biology\",\"volume\":\"79 Pt 1\",\"pages\":\"10-21\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9815095/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Crystallographica. Section D, Structural Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1107/S2059798322011676\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Crystallographica. Section D, Structural Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1107/S2059798322011676","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Protein model refinement for cryo-EM maps using AlphaFold2 and the DAQ score.
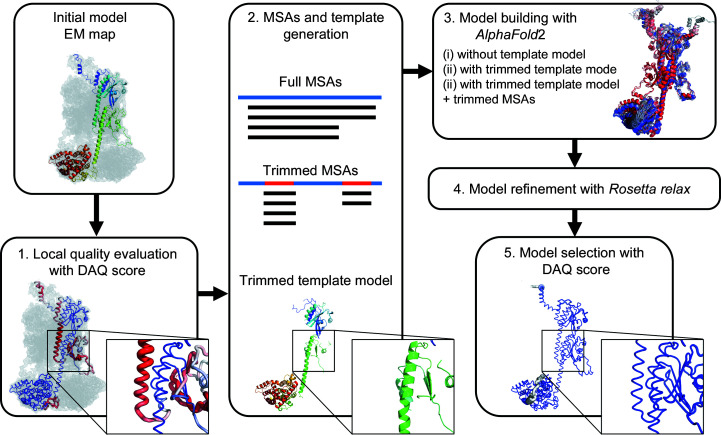
As more protein structure models have been determined from cryogenic electron microscopy (cryo-EM) density maps, establishing how to evaluate the model accuracy and how to correct models in cases where they contain errors is becoming crucial to ensure the quality of the structural models deposited in the public database, the PDB. Here, a new protocol is presented for evaluating a protein model built from a cryo-EM map and applying local structure refinement in the case where the model has potential errors. Firstly, model evaluation is performed using a deep-learning-based model-local map assessment score, DAQ, that has recently been developed. The subsequent local refinement is performed by a modified AlphaFold2 procedure, in which a trimmed template model and a trimmed multiple sequence alignment are provided as input to control which structure regions to refine while leaving other more confident regions of the model intact. A benchmark study showed that this protocol, DAQ-refine, consistently improves low-quality regions of the initial models. Among 18 refined models generated for an initial structure, DAQ shows a high correlation with model quality and can identify the best accurate model for most of the tested cases. The improvements obtained by DAQ-refine were on average larger than other existing methods.
期刊介绍:
Acta Crystallographica Section D welcomes the submission of articles covering any aspect of structural biology, with a particular emphasis on the structures of biological macromolecules or the methods used to determine them.
Reports on new structures of biological importance may address the smallest macromolecules to the largest complex molecular machines. These structures may have been determined using any structural biology technique including crystallography, NMR, cryoEM and/or other techniques. The key criterion is that such articles must present significant new insights into biological, chemical or medical sciences. The inclusion of complementary data that support the conclusions drawn from the structural studies (such as binding studies, mass spectrometry, enzyme assays, or analysis of mutants or other modified forms of biological macromolecule) is encouraged.
Methods articles may include new approaches to any aspect of biological structure determination or structure analysis but will only be accepted where they focus on new methods that are demonstrated to be of general applicability and importance to structural biology. Articles describing particularly difficult problems in structural biology are also welcomed, if the analysis would provide useful insights to others facing similar problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


